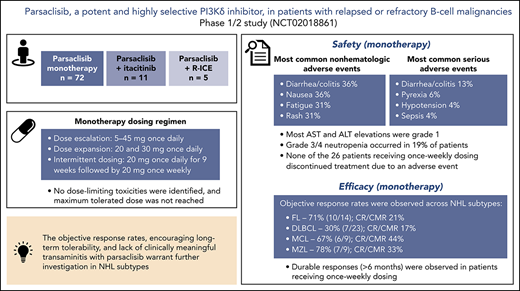
Key Points
Parsaclisib is a potent, highly selective, next-generation PI3Kδ inhibitor that is not associated with clinically meaningful transaminitis.
The objective response rates and encouraging long-term tolerability observed in this study warrant further investigation in NHL subtypes.

Abstract
This phase 1/2 study assessed parsaclisib (INCB050465), a next-generation, potent, and highly selective phosphatidylinositol 3-kinase δ (PI3Kδ) inhibitor, in patients with relapsed or refractory B-cell malignancies, alone or in combination with a Janus kinase 1 inhibitor (itacitinib) or chemotherapy (rituximab, ifosfamide, carboplatin, and etoposide). Seventy-two patients received parsaclisib monotherapy (5-45 mg once daily). Expansion doses were 20 and 30 mg once daily; intermittent dosing at 20 mg (once daily for 9 weeks, then once weekly) was explored. No dose-limiting toxicities were identified, and maximum tolerated dose was not reached. Most common nonhematologic treatment-emergent adverse events (TEAEs) were diarrhea/colitis (36%), nausea (36%), fatigue (31%), and rash (31%). Grade 3/4 neutropenia occurred in 19% of patients. Serious TEAEs (>2 patients) were diarrhea/colitis (n = 9), pyrexia (n = 4), hypotension (n = 3), and sepsis (n = 3). Aspartate and alanine transaminase elevations occurring before treatment discontinuation were grade 1, except 1 grade 3 event each, secondary to sepsis. Two patients experienced 3 fatal parsaclisib-unrelated TEAEs (respiratory failure; respiratory failure and sepsis). In non-Hodgkin lymphoma (NHL), objective response rates to monotherapy were 71% in follicular lymphoma, 78% in marginal zone lymphoma, 67% in mantle cell lymphoma, and 30% in diffuse large B-cell lymphoma; 93% of responses occurred at first assessment (∼9 weeks). Parsaclisib has demonstrated antitumor activity in relapsed or refractory B-cell NHL with the potential for improved long-term patient outcomes. Phase 2 studies in relapsed or refractory B-cell NHL subtypes are ongoing. This trial was registered at www.clinicaltrials.gov as #NCT02018861.
Introduction
Constitutive signaling through B-cell receptors plays a critical role in the pathogenesis of human B-cell malignancies1 and leads to downstream activation of class I phosphatidylinositol 3-kinases (PI3Ks).2,3 Class I PI3Ks are heterodimeric lipid kinases composed of a regulatory (p85 or p101) and a catalytic (p110) subunit.4 Each of the 4 tissue-specific p110 subunit isoforms (class IA: α, β, and δ; class IB: γ) confers unique physiologic functions on the corresponding PI3K isoforms.5-9 The PI3Kδ isoform functions as a critical node in signaling networks that regulate B-cell growth and survival, and its aberrant activation is a key event in malignant transformation of B cells.10,11 Substantial interconnectivity exists between B-cell receptors and PI3Kδ-mediated signaling networks and other networks important for regulating B-cell survival and proliferation, including the Janus kinase (JAK)–signal transducer and activator of transcription pathway,12,13 suggesting potential additive or synergistic therapeutic effects in B-cell malignancies.
The 5-year overall survival rate for patients with relapsed follicular lymphoma (FL), the most common indolent non-Hodgkin lymphoma (NHL) subtype, is only 50%.14 Prognosis is worse for patients with relapsed aggressive NHL subtypes, with a median survival of 3.6 and 4.4 months among patients with relapsed diffuse large B-cell lymphoma (DLBCL) who had failed first-line and second-line salvage regimens, respectively.15 Current guidelines for the treatment of relapsed B-cell NHL differ according to subtype and include immunochemotherapy, radioimmunotherapy, targeted therapies with small-molecule kinase inhibitors, or immunomodulatory therapies (including chimeric antigen receptor T-cell therapy).16-20 In addition to systemic therapy, autologous or allogeneic stem cell transplant (SCT) is often used to treat patients with relapsed B-cell NHL and is considered curative for certain patients.21-25
For patients with relapsed or refractory disease, the PI3K inhibitor class has shown promise, but clinical use has been limited by toxicities.26-33 Parsaclisib (INCB050465) is a potent and highly selective next-generation PI3Kδ inhibitor (≥19 000-fold selectivity for PI3Kδ over other PI3K class I isoforms; whole-blood half-maximal inhibitory concentration [IC50] = 10 nM; 90% of maximal inhibitory concentration [IC90] = 77 nM).34,35 The structure of parsaclisib differs fundamentally from first-generation PI3Kδ inhibitors that have entered the clinic. Specifically, parsaclisib comprises a monocyclic scaffold with a pyrazolopyrimidine substituent compared with a bicyclic scaffold with a purine substituent for first-generation PI3Kδ inhibitors.34 The hepatotoxicity observed in the clinic with first-generation PI3Kδ inhibitors is believed to be an off-target effect associated with these highly conserved structural features, and thus, the distinct structure of parsaclisib should limit these off-target toxicities. Accordingly, preclinical toxicology studies with parsaclisib demonstrated no hepatotoxicity at exposures that exceeded IC90 coverage by more than 10-fold.34 In primary cell-based assays, parsaclisib potently inhibited proliferation of malignant human B cells with mean IC50 values lower than 1 nM.34 Single-agent parsaclisib also inhibited tumor growth in DLBCL xenograft models, and the antitumor effect was enhanced when combined with JAK1- and pan-Proviral Integration site of Moloney murine leukemia virus-selective kinase inhibitors, as well as inhibitors of epigenetic regulators (eg, bromo- and extraterminal domain; lysine-specific histone demethylase 1A).36
The objective of this study was to assess the safety, tolerability, preliminary efficacy, pharmacokinetics, and pharmacodynamics of parsaclisib, alone or combined with the JAK1 inhibitor, itacitinib, or with immunochemotherapy, in patients with relapsed or refractory B-cell malignancies.
Methods
Study design and patients
This phase 1/2, open-label, dose-escalation, and dose-expansion study (CITADEL-101) was conducted in multiple parts: dose escalation of parsaclisib monotherapy (part 1) followed by cohort expansion (part 3); parsaclisib plus itacitinib dose escalation (part 2) followed by cohort expansion (part 3); and parsaclisib plus R-ICE (rituximab plus ifosfamide, carboplatin, and etoposide) dose evaluation and expansion (part 6) (Figure 1). Parts 4 and 5 were removed by a later protocol amendment (19 May 2016; supplemental Data, available on the Blood Web site). Eligible patients (age ≥18 years) had diagnosed lymphoid malignancies of B-cell origin, including indolent or aggressive B-cell NHL malignancies, transformed NHL histologies, and HL, that had relapsed or were refractory to prior standard therapy. Patients with Burkitt lymphoma and precursor B-lymphoblastic leukemia/lymphoma were excluded. Eligible patients had Eastern Cooperative Oncology Group performance status of 1 or less (dose escalation) or 2 or less (dose expansion); adequate cardiac, liver, and kidney function; life expectancy at least 12 weeks; received at least 1 prior treatment regimen; and had not responded to or were not candidates for hematopoietic (H)SCT or other potentially curative therapy.
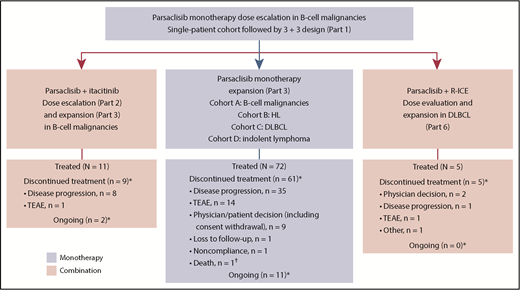
Study design and patient disposition. Parts 4 and 5 were removed by a later protocol amendment (19 May 2016). HL, Hodgkin lymphoma; TEAE, treatment-emergent adverse event. *As of data cutoff (18 August 2017). †Because of septic shock and respiratory failure.
Study design and patient disposition. Parts 4 and 5 were removed by a later protocol amendment (19 May 2016). HL, Hodgkin lymphoma; TEAE, treatment-emergent adverse event. *As of data cutoff (18 August 2017). †Because of septic shock and respiratory failure.
Patients were excluded if they had a history of untreated, symptomatic, or unstable brain metastases; spinal cord compression; lymphoma involving the central nervous system (permitted in the parsaclisib monotherapy expansion cohort A [B-cell malignancies, Figure 1] and parsaclisib plus itacitinib expansion cohort); or received allogeneic HSCT within 6 months or autologous HSCT within 3 months of enrollment. Prior treatment with PI3Kδ inhibitors was excluded for parsaclisib monotherapy cohorts unless approved by the medical monitor, and prior JAK inhibitors were excluded for parsaclisib plus itacitinib cohorts. Additional exclusion criteria are provided in the supplemental Data.
The study was conducted at 13 US sites in accordance with the study protocol, Declaration of Helsinki, Good Clinical Practice, International Conference on Harmonisation, and applicable regulatory requirements. All patients provided written, informed consent before study participation. The study protocol was approved by the respective institutional review boards or independent ethics committees. D.J.D., L.Z., and X.C. analyzed the data, and all authors had access to study data. This trial was registered at www.clinicaltrials.gov as #NCT02018861.
Dosing
For monotherapy, parsaclisib was self-administered orally once daily in a fasted state (except for patients participating in an optional food-effect cohort) until an amendment on 2 August 2017, allowed administration without regard to food. Treatment continued until unacceptable toxicity or disease progression. Dose escalation (Table 1) was planned as an initial single-patient cohort treated with 5 mg once daily, and subsequent cohorts were enrolled using a 3 + 3 design, to identify any DLT (defined in supplemental Table 1) within the 21-day observation period. If DLTs occurred in more than 1 of the first 3 patients or the total cohort of 6 patients, then the MTD was deemed to be exceeded and the next lower tolerable dose level was defined as the MTD. All doses up to 45 mg once daily were well tolerated over the DLT observation period of 21 days, and therefore, no MTD was identified. However, incidences of colitis were observed over the longer term (>3 months), prompting a change in dosing regimen. On 29 November 2016, a protocol amendment modified the dosing schedule to daily dosing followed by weekly dosing, based on a pharmacokinetic simulation (Discussion). Patients enrolled from this date received parsaclisib 20 mg once daily for the first 9 weeks, followed by parsaclisib 20 mg once weekly; patients who had already received parsaclisib 20 mg once daily for at least 9 weeks were switched to the once-weekly schedule. As of 3 May 2017, per protocol amendment, ongoing and newly enrolled patients were required to take prophylactic treatment for Pneumocystis jiroveci pneumonia (PJP). See supplemental Data for details of parsaclisib plus itacitinib or R-ICE combination dosing.
Assessments
Patients were assessed on days 1, 8, and 15 of cycle 1 and day 1 (±3) of each subsequent treatment cycle. A schedule of key study assessments is shown in supplemental Table 2.
The primary end point was safety and tolerability of parsaclisib monotherapy, parsaclisib plus itacitinib, or parsaclisib plus R-ICE, as assessed by summary of TEAEs, clinical laboratory assessments, physical examination results, and 12-lead electrocardiograms. The severity of TEAEs was assessed using Common Terminology Criteria for Adverse Events version 4.03.
The secondary end points were efficacy and pharmacokinetics. Efficacy was measured by best overall response rate (ORR), defined as the proportion of patients achieving a partial response or a complete response (CR). Response was evaluated (per investigator's assessment) every 9 weeks, based on the Lugano Classification of lymphoma response criteria for HL and NHL,37 the International Working Group on Chronic Lymphocytic Leukemia criteria for chronic lymphocytic leukemia (CLL),38,39 and the VIth International Workshop on Waldenström macroglobulinemia response assessment for Waldenström macroglobulinemia (WM).40 If the patient’s response to treatment was assessed by positron emission tomography (PET) and computed tomography (CT)/magnetic resonance imaging, the PET result was used for response end points; the CT/magnetic resonance imaging result was used for an assessment if the PET result was not available for the assessment. PET was required for DLBCL and HL and was optional for all other patients.
Exploratory end points included: duration of response (duration of time from first response to death or disease progression, whichever occurred first), progression-free survival (duration of time from first study dose to death or disease progression, whichever occurred first), population pharmacokinetics, and pharmacodynamic relationship between PI3Kδ inhibition (as determined by an ex vivo phosphorylated AKT [pAKT] assay [for additional details, see supplemental Data, “Ex vivo pAKT assay”]).
Statistical methods
Safety, tolerability, and efficacy analyses were performed on the safety/intent-to-treat population, which included all enrolled patients who received at least 1 dose of parsaclisib, itacitinib, or any of the R-ICE components. Pharmacokinetic and pharmacodynamic analyses included all patients in the safety/intent-to-treat population with available pharmacokinetic/pharmacodynamic data. All statistical analyses were exploratory in nature and were summarized using descriptive statistics. ORRs were estimated with 95% confidence intervals (CIs) calculated based on the exact method for binomial distributions. Kaplan-Meier estimates of median duration of response and progression-free survival were presented with their respective 95% CIs.
Results
This analysis includes data from patients enrolled into parsaclisib monotherapy dose-escalation (5-45 mg once daily) and dose-expansion (20 mg and 30 mg once daily) cohorts. Data obtained from patients receiving parsaclisib plus itacitinib or R-ICE are also presented.
Parsaclisib monotherapy
Patients
Between 1 April 2015, and 12 May 2017, 72 patients were enrolled and treated with parsaclisib monotherapy (median age, 66 years; ≥3 prior systemic therapies, 54%; prior HSCT, 29%; Table 2). The median (range) duration of treatment was 4.0 (0.2-22.7) months. At the data cutoff of 18 August 2017, all but 11 patients had discontinued study treatment, primarily for disease progression (n = 35 [49%]) and TEAEs (n = 14 [19%]; Figure 1).
Dose escalation and cohort expansion
The number of patients per dose level during parsaclisib monotherapy dose escalation and expansion is shown in Table 1. No DLTs were identified, the MTD was not reached, and the 20-mg and 30-mg once-daily doses were expanded.
Pharmacokinetics and pharmacodynamics of parsaclisib
All doses tested remained above the IC90 for target inhibition throughout the dosing interval (Figure 2A); maximum concentration (Cmax) and lowest concentration that a drug reaches before the next dose is administered resulting from a 20-mg once-daily dose of parsaclisib were 16-fold and 2-fold above the IC90 for pAKT inhibition, respectively. A pharmacokinetic simulation conducted to support selection of a 20-mg once-weekly dosing schedule estimated that serum parsaclisib levels would exceed the IC90 for target inhibition for ∼36 hours and would have minimal to no inhibition for approximately half the dosing interval (supplemental Figure 1). After multiple-dose administration of parsaclisib alone, parsaclisib attained peak plasma concentrations rapidly, with a median time to Cmax of 0.5 to 1 hour. Subsequently, parsaclisib plasma concentrations declined in a monophasic fashion with a mean terminal phase disposition half-life of 8.6 to 11.5 hours. Dose proportionality appeared to be observed between 5 and 45 mg once daily at steady state (Figure 2B).
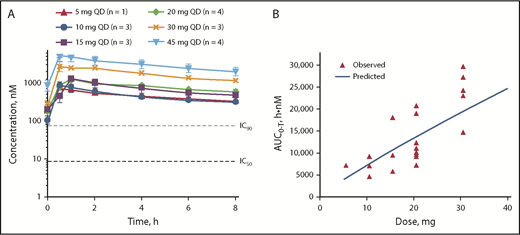
Pharmacokinetics of parsaclisib at steady state. (A) Pharmacokinetics of parsaclisib dosing. (B) Dose-response relationship of parsaclisib area under the curve (AUC). AUC0-T, area under the plasma concentration-time curve; QD, once daily.
Pharmacokinetics of parsaclisib at steady state. (A) Pharmacokinetics of parsaclisib dosing. (B) Dose-response relationship of parsaclisib area under the curve (AUC). AUC0-T, area under the plasma concentration-time curve; QD, once daily.
Food effect was analyzed in 12 patients. Although co-administration of parsaclisib with a high-fat meal decreased Cmax by 42% compared with the fasted state, it had a modest effect on AUC (10% decrease). The fed vs fasted geometric mean ratio for AUC was 0.90 (90% CI, 0.68-1.18).
Near-maximal PI3Kδ inhibition was observed at all parsaclisib doses for up to 6 hours postdose, as measured by effects on pAKT levels in whole blood (supplemental Figure 2).
Safety and tolerability
The most common (≥30%) any-grade nonhematologic TEAEs in all patients were diarrhea/colitis (36%), nausea (36%), fatigue (31%), and rash (31%) (Table 3). New or worsening of any-grade and grade 3/4 neutropenia occurred in 32 patients (44%) and 14 patients (19%), respectively (Table 4). Serious adverse events (SAEs) experienced by more than 2 patients were diarrhea/colitis (n = 9 [13%]), pyrexia (n = 4 [6%]), hypotension (n = 3 [4%]), and sepsis (n = 3 [4%]). Any-grade SAEs of infections and infestations (System Organ Class) occurred in 11 patients (15%), including pneumonia in 2 patients (3%).
Among TEAEs of interest, the median (range) time to onset of grade 3/4 diarrhea/colitis was 5.7 (1.6-14.9) months, and median (range) time to onset of grade 3/4 rash was 2.9 (1.5-9.3) months. Aspartate transaminase (AST) and alanine transaminase (ALT) elevations occurred in 21 (29%) and 20 (28%) patients, respectively; all events were grade 1 with the exception of 4 events in 2 patients (1 patient with 1 event each of grade 3 AST elevation and grade 2 ALT elevation, both occurring more than 30 days after the last dose of parsaclisib; 1 patient with 1 event each of grade 3 AST elevation and ALT elevation considered secondary to sepsis). Bilirubin elevation occurred in 8 patients (11%); all events were grade 2 or lower with the exception of 1 grade 3 event that occurred more than 30 days after the date of the last dose. Changes in AST and ALT levels over time are shown in supplemental Figure 3. One patient (1%) each experienced grade 3 pneumonitis and grade 3 cytomegalovirus colitis, and 7 patients (10%) experienced pneumonia (grade 3, n = 3 [4%]); none of the cases was considered to be related to parsaclisib. There were no TEAEs of PJP or bowel perforation. Five patients (7%) experienced hypertension (all grade 1/2); 7 (10%) experienced hyperglycemia (all grade 1/2 except 1 grade 3).
Any-grade TEAEs led to parsaclisib dose interruption in 30 patients (42%), dose reduction in 4 patients (6%), and treatment discontinuation in 14 patients (19%). The most common nonhematologic TEAEs leading to parsaclisib dose interruption were diarrhea/colitis (11/72 [15%]) and pyrexia (4/72 [6%]). TEAEs leading to death occurred in 2 patients (respiratory failure; and respiratory failure and sepsis); both occurred in the setting of disease progression, and none was deemed related to parsaclisib.
Long-term tolerability and dosing
Before implementation of the once-weekly dosing schedule, 9 (29%) of 31 patients with DLBCL, FL, MCL, and MZL receiving parsaclisib monotherapy had discontinued because of TEAEs, most commonly (n ≥ 2) diarrhea/colitis (n = 3 [10%]) or rash (n = 2 [6%]); however, no patient had discontinued because of treatment-related AEs during the first 9 weeks of treatment. As of the data cutoff date, 26 (36%) of 72 enrolled patients had received once-weekly dosing of parsaclisib (after receiving once-daily dosing for at least 9 weeks; 15 patients switched to once-weekly dosing between weeks 8 and 10) for a median (range) of 2.5 (1.2-9.6) months and a total of 105 patient-months; 46 patients (64%) received once-daily dosing only; all patients completed the first 9 weeks of once-daily dosing. Four of 26 patients initiated the once-weekly schedule at lower than a 20-mg dose because of previous dose reductions (1 at 5 mg, 3 at 10 mg). Among the 26 patients who received once-weekly dosing, 8 (31%) had dose interruptions (TEAEs leading to dose interruption in >1 patient were diarrhea and neutropenia [n = 2 each]), 1 (4%) had a dose reduction (because of rash pustular), and none discontinued treatment because of a TEAE. During the once-weekly dosing period, 4 patients (15%) experienced 6 SAEs (abdominal pain, diarrhea, pyrexia, bronchitis, sepsis, and dehydration; n = 1 each). No grade 4 nonhematologic TEAEs were reported, and 2 patients had grade 3 diarrhea and rash (n = 1 [4%] each); both events occurred shortly after the switch from once-daily dosing. No new or worsening grade 4 neutropenia was reported, and 1 patient experienced new or worsening grade 4 thrombocytopenia.
Efficacy
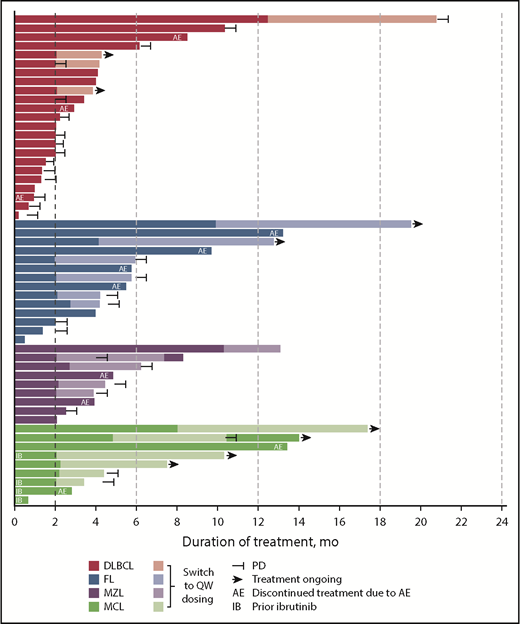
ORR among evaluable patients varied by disease type (Table 5); 7 (30%) of 23 patients with DLBCL, 10 (71%) of 14 patients with FL, 7 (78%) of 9 patients with MZL, and 6 (67%) of 9 patients with MCL achieved an objective response. The CR/CMR rates for these 4 subtypes were 4/23 (17%), 3/14 (21%), 3/9 (33%), and 4/9 (44%), respectively. Best percentage change from baseline in target lesion size for patients with evaluable lesion size at baseline and postbaseline is shown in Figure 3. Among patients with these tumor types, the vast majority (93%) of responses occurred by the first assessment (∼9 weeks; supplemental Figure 4). Durable responses (>6 months) were observed among patients receiving once-weekly dosing, and several patients remained on the once-weekly schedule for at least 8 months (Figure 4), including 2 who were administered 10 mg once weekly. The median duration of response was 13.5 months (95% CI, 8.3-18.8) for DLBCL and 4.4 months (95% CI, 2.1-not evaluable) for MZL, and was not reached for FL or MCL.
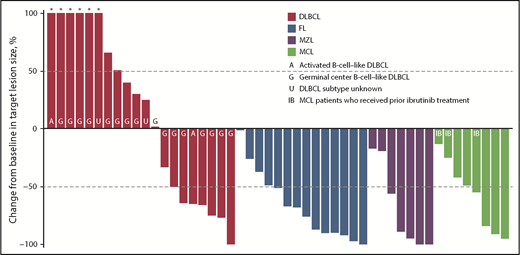
Monotherapy best percentage change from baseline in target lesion size. Data for 3 patients with DLBCL, 2 with MZL, and 1 with MCL are not shown because of nonmeasurable disease at baseline or no valid postbaseline target lesion measurements. *Best percentage change from baseline in target lesion size more than 100%.
Monotherapy best percentage change from baseline in target lesion size. Data for 3 patients with DLBCL, 2 with MZL, and 1 with MCL are not shown because of nonmeasurable disease at baseline or no valid postbaseline target lesion measurements. *Best percentage change from baseline in target lesion size more than 100%.
Parsaclisib combination therapy
Parsaclisib combined with itacitinib
Eleven patients were enrolled and treated with parsaclisib 20 mg (n = 8) or 30 mg (n = 3) once daily plus itacitinib 300 mg once daily (Figure 1). No DLTs were observed. The median (range) duration of both parsaclisib and itacitinib treatment was 2.1 (1.0-10.3) months. At data cutoff, treatment was ongoing in 2 patients (18%); 8 patients (73%) discontinued treatment because of disease progression and 1 (9%) because of a TEAE (see supplemental Figure 5 for pharmacokinetics).
Nine patients (82%) experienced TEAEs, and 5 patients (45%) experienced grade 3/4 TEAEs: grade 3 spinal cord compression, intractable pain, and elevated alkaline phosphatase (n = 1); grade 3 worsening hyperlipidemia (n = 1); grade 3 hypertension (n = 1); grade 3 diarrhea and dehydration (n = 1); and grade 3 hypertension and grade 4 hypercalcemia (n = 1). One patient had a new/worsening grade 4 hematologic laboratory abnormality (thrombocytopenia). Two patients (18%) had SAEs: grade 3 dehydration and grade 4 hypercalcemia (n = 1 each). TEAEs leading to dose interruption or reduction of parsaclisib and itacitinib occurred in 2 patients (18%) and 1 patient (9%), respectively; no patient had a TEAE leading to dose reduction or death.
Among evaluable patients treated with parsaclisib plus itacitinib, best overall response of CR/CMR was achieved by patients with CLL (1/1) and FL (1/1); best overall response of PMR was achieved by patients with MCL (1/1) and classic HL (1/2). All 6 patients with DLBCL and 1 of 2 patients with classic HL had best overall response of PD/PMD.
Parsaclisib combined with R-ICE
Five patients with DLBCL were treated with parsaclisib 15 mg (n = 4) or 20 mg (n = 1) once daily plus R-ICE (Figure 1). No DLTs were observed. The median (range) duration of parsaclisib treatment was 2.3 (1.5-3.7) months; 4 patients received 3 cycles of R-ICE each, and 1 patient received 2 cycles of R-ICE. As of the data cutoff date, all patients have discontinued treatment because of physician decision (n = 2), PD, TEAEs, and other (n = 1 each; see supplemental Figure 5B for pharmacokinetics).
Three patients reported new/worsening grade 4 thrombocytopenia, and 2 patients reported new/worsening grade 4 neutropenia. Two patients experienced SAEs, of whom 1 experienced grade 2 atrial flutter and atrial fibrillation and grade 3 dyspnea, neutropenia, and febrile neutropenia (all related to parsaclisib and R-ICE). Three patients had TEAEs leading to dose interruption of parsaclisib and R-ICE, 1 patient had a TEAE leading to dose interruption of only parsaclisib, and 1 patient had TEAEs leading to discontinuation of parsaclisib and R-ICE; no patient had a TEAE leading to dose reduction or death.
Three patients achieved a CMR (1 of 3 patients indicated a desire to proceed to SCT), and 2 patients had SD as best response.
Discussion
In this phase 1/2, open-label, dose-escalation, and dose-expansion study, the pharmacokinetics, pharmacodynamics, safety, and efficacy of parsaclisib were evaluated in patients with relapsed or refractory B-cell malignancies. The pharmacokinetic characteristics of parsaclisib combined with the absence of DLTs permitted exposure levels that exceeded the IC90 throughout the once-daily dosing interval at all doses tested. In addition, the results of a food-effect analysis indicated that parsaclisib may be dosed without regard to timing of meals. The near-maximal inhibition of pAKT in the ex vivo pharmacodynamic assay was consistent with exposure data.
Parsaclisib demonstrated a differentiated safety profile relative to first-generation PI3Kδ inhibitors (eg, idelalisib and duvelisib),29,41 most notably by the near absence of grade 2 or above transaminitis. Although we cannot definitively exclude the potential of parsaclisib to cause clinically meaningful hepatotoxicity, the molecular structure was designed to limit off-target hepatotoxic effects observed with first-generation PI3Kδ inhibitors. Accordingly, there were no Hy’s Law cases, and no patient discontinued treatment because of hepatotoxicity. The exact cause of transaminitis observed with first-generation PI3Kδ inhibitors is not known, but we believe it is associated with their conserved structural features. Increased ALT/AST was observed in 50% (19% grade 3/4)/41% (12% grade 3/4) of patients with indolent NHL treated with idelalisib,42 and in 40% (8% ≥ grade 3)/37% (6% ≥ grade 3) of patients with B-cell malignancies receiving duvelisib.43 We also report no clinically meaningful hypertension or hyperglycemia (11/12 cases were grade 1/2 and likely related to baseline medical status), which we believe to be the result of the high selectivity of parsaclisib for the δ isoform of PI3K and near absence of α isoform inhibition at therapeutic doses. High rates of hypertension (35%; 27% grade 3) and hyperglycemia (54%; 39% grade 3/4) have been reported with the pan-PI3K inhibitor copanlisib, which inhibits the α and δ isoforms and is approved in the United States as monotherapy for the treatment of relapsed FL.26 Although no PJP was observed, a protocol amendment made PJP prophylaxis mandatory partway through the study in response to a PJP infection reported in a different phase 1 study that combined parsaclisib with pembrolizumab (Incyte Corporation unpublished data, 2 August 2017).
Diarrhea/colitis and rash, toxicities common to PI3K inhibitors, were 2 of the most frequent toxicities observed in this study; nevertheless, the majority of diarrhea events were of grade 1 or 2. The majority of grade 3/4 diarrhea/colitis and rash occurred after the first disease assessment scheduled at 9 weeks; overall, any-grade diarrhea/colitis or rash led to treatment discontinuation in 9 (13%) of 72 patients, including 3 patients with diarrhea/colitis who redeveloped diarrhea/colitis on reinitiating treatment at the same dose. Grade 4 neutropenia (6%) was observed only during once-daily dosing (earliest occurring after 21 days of treatment). The safety profile associated with the 20-mg once-daily dose was generally consistent with that observed for all doses combined and with that observed for each individual dose level (data not shown), although relatively few patients were dosed at levels below 20 mg and above 30 mg once daily.
Long-term dosing of parsaclisib identified late-onset TEAEs that led to treatment discontinuation. To improve the long-term tolerability of parsaclisib while maintaining a high response rate, a modified dosing regimen was implemented (20 mg once daily for 9 weeks followed by 20 mg once weekly). The period of daily dosing was maintained to preserve the potential for rapid onset of response (at the time of implementation, all 11 patients with NHL who were administered 20 mg once daily had achieved an objective response, 10 of which occurred at the first disease assessment). The intermittent portion of the dosing regimen, which was implemented to decrease the incidence of late-onset TEAEs, was based on comparative pharmacokinetic/pharmacodynamic simulation with copanlisib.33 The 20-mg once-weekly regimen is anticipated to provide similar pAKT inhibition, as expected from once-weekly dosing of copanlisib. Encouragingly, none of the 26 patients who received once-weekly dosing discontinued study treatment because of TEAEs, according to data accrued during a 105 patient-month period. This observation, combined with the absence of grade 4 nonhematologic TEAEs and grade 4 neutropenia, and a low rate of grade 3 diarrhea and rash (n = 1 each), suggests that the once-weekly schedule is better tolerated than the continuous once-daily schedule using the doses evaluated herein.
Parsaclisib monotherapy effected a high rate of rapid and durable objective responses in patients with heavily pretreated relapsed or refractory B-cell NHL at doses ranging from 10 to 45 mg once daily. ORR across all subtypes ranged from 30% to 78% (DLBCL, 30%; MCL, 67%; FL, 71%; MZL, 78%). Among patients with DLBCL, substantially more patients with GCB subtype (n = 19) were enrolled relative to those with ABC subtype (n = 2). These small, disproportionate numbers are insufficient to determine whether parsaclisib is more active in 1 subtype than the other. Responses in patients with MCL are of particular interest, given that no PI3Kδ inhibitors are currently approved in this indication. Anecdotally, objective responses were observed in 2 of the 4 patients with MCL previously treated with ibrutinib; 1 of these responses was ongoing at approximately 6 months at data cutoff. Durable responses were observed in all 4 of these NHL subtypes, including ∼19 months in DLBCL, ∼17 months in FL, ∼10 months in MZL, and ≥14 months in MCL, all of which included once-weekly dosing. Responses were ongoing at data cutoff in 3 of the 4 NHL subtypes. Taken together, these data suggest that once-weekly dosing of parsaclisib can enable durable responses in patients with B-cell NHL.
The pharmacokinetic and safety data from the combination studies show that parsaclisib can be safely combined with itacitinib (300 mg once daily). Similarly, parsaclisib plus R-ICE also appears to be tolerated. Additional studies are required to further evaluate the safety of parsaclisib plus R-ICE and the potential for increased efficacy for each of the combinations.
In conclusion, parsaclisib demonstrated preliminary antitumor activity with rapid responses and a differentiated toxicity profile in patients with relapsed or refractory B-cell malignancies. The molecular structure is hypothesized to minimize the frequency of clinically meaningful hepatotoxicity, and the modified dosing regimen explored here appeared to improve the long-term tolerability of parsaclisib and enable durable responses in several patients. Nevertheless, the optimal dosing regimen was not established, and investigations into this and other dosing regimens are ongoing. The efficacy, safety, pharmacokinetic, and pharmacodynamic data presented here support further investigation of parsaclisib monotherapy in ongoing phase 2 studies in select NHL subtypes (DLBCL [NCT02998476], FL [NCT03126019], MZL [NCT03144674], and MCL [NCT03235544]). As the most promising opportunities for PI3Kδ inhibition may be in combination with standard-of-care agents, phase 1 studies exploring combination strategies in patients with NHL (NCT03039114; NCT03424122) are also underway.
Presented in poster/oral presentation form (earlier/preliminary data cuts) at the American Association for Cancer Research Annual Meeting, New Orleans, LA, 16-20 April 2016; the 58th annual meeting of the American Society of Hematology, San Diego, CA, 3-6 December 2016; the 2017 Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, 2-6 June 2017; the 14th International Conference on Malignant Lymphoma, Lugano, Switzerland, 14-17 June 2017; the 22nd Congress of the European Hematology Association, Madrid, Spain, 22-25 June 2017; and the 59th annual meeting of the American Society of Hematology, Atlanta, GA, 9-12 December 2017.
Access to individual patient level-data is not available for this study.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients who participated in this study and the investigators and teams who conducted the study. The authors acknowledge the contributions of Steven Shi (Incyte Corporation), who provided statistical programming support. Medical writing assistance was provided by Karolina Rzeniewicz (Evidence Scientific Solutions Inc., Philadelphia, PA) and funded by Incyte Corporation.
This study was supported by Incyte Corporation (Wilmington, DE).
Authorship
Contribution: A.F.-T., R.R., A.Y., M.S.W., W.J.E., P.C., M.G., L.A., C.E., J.C., D.P., S.I., D.J.D., L.Z., X.C., F.D., and T.J.P. were involved in designing and/or conduct of the study; A.F.-T., R.R., A.Y., M.S.W., W.J.E., P.C., M.G., L.A., C.E., J.C., D.P., S.I., and T.J.P. acquired the data; D.J.D., L.Z., and X.C. analyzed the data; and all authors interpreted the data, drafted the manuscript and/or revised it critically, and approved the final draft.
Conflict-of-interest disclosure: A.F.-T. served on the speakers’ bureau for Seattle Genetics, had a consulting or advisory role with Incyte, and received research funding to the institution from Daiichi Sankyo, Genentech/Roche, Gilead Sciences, Immunomedics, Novartis, Oncothyreon, Pfizer, Seattle Genetics, Syndax, and TRACON Pharma. R.R. had a consulting or advisory role with Curis, Gilead Sciences, and Seattle Genetics and received research funding from Janssen, Merck, and Pharmacyclics. A.Y. served on the speakers’ bureau for Incyte, Novartis, and Seattle Genetics and held a consulting or advisory role for CTI, Incyte, Pfizer, Sandoz, and Seattle Genetics. M.S.W. served on the speakers’ bureau for AstraZeneca, Celgene, and Millennium. P.C. held a consulting or advisory role with Genentech and Kite Pharma. M.G. holds stock and other ownership interests in COTA (Cancer Outcomes Tracking and Analysis); had a consulting or advisory role for Bayer, Karyopharm Therapeutics, and Lilly; served on the speakers’ bureau for Bristol-Myers Squibb, Foundation One Inc, and Merck; and received research funding to the institution from AbbVie, Acceleron Pharma, Bristol-Myers Squibb, Daiichi Sankyo, EMD Serono, Esanex, Gilead Sciences, Incyte, Karyopharm Therapeutics, Lilly, Loxo, MedImmune, Merck, Novartis, Rexahn Pharmaceuticals, and TG Therapeutics. L.A. held a consulting or advisory role with Novartis; served on the speakers’ bureau for Bristol-Myers Squibb, Celgene, Gilead Sciences, Millennium, Novartis, and Teva; and received research funding from Astellas Medivation, Cellerant, Incyte, Pfizer, Teva, and Unum Therapeutics. C.E. is an employee of Texas Oncology, P.A., served on the speakers’ bureau for Kite Pharma and Seattle Genetics, and has a membership with an entity’s board of directors or advisory committees at the Vitruvian Institute for Medical Advancements. D.P. had a consulting or advisory role with Genentech, MorphoSys, and Spectrum Pharmaceuticals and received research funding to the institution from Merck. D.J.D., L.Z., X.C., and F.D. are employees of/own stock in Incyte. T.J.P. had a consulting or advisory role with Pharmacyclics and Seattle Genetics, received research funding from Incyte, and received travel and accommodations expenses from Incyte. The remaining authors declare no competing financial interests.
Correspondence: Andres Forero-Torres, 3101 Western Ave, Suite 600, Seattle, WA 98121; e-mail: andresforeromd@gmail.com.