

Abstract
A vast amount of work has been dedicated to the effects of shear flow and cytokines on leukocyte transmigration. However, no studies have explored the effects of substrate stiffness on transmigration. Here, we investigated important aspects of endothelial cell contraction-mediated neutrophil transmigration using an in vitro model of the vascular endothelium. We modeled blood vessels of varying mechanical properties using fibronectin-coated polyacrylamide gels of varying physiologic stiffness, plated with human umbilical vein endothelial cell (HUVEC) monolayers, which were activated with tumor necrosis factor-α. Interestingly, neutrophil transmigration increased with increasing substrate stiffness below the endothelium. HUVEC intercellular adhesion molecule-1 expression, stiffness, cytoskeletal arrangement, morphology, and cell-substrate adhesion could not account for the dependence of transmigration on HUVEC substrate stiffness. We also explored the role of cell contraction and observed that large holes formed in endothelium on stiff substrates several minutes after neutrophil transmigration reached a maximum. Further, suppression of contraction through inhibition of myosin light chain kinase normalized the effects of substrate stiffness by reducing transmigration and eliminating hole formation in HUVECs on stiff substrates. These results provide strong evidence that neutrophil transmigration is regulated by myosin light chain kinase-mediated endothelial cell contraction and that this event depends on subendothelial cell matrix stiffness.
Introduction
Leukocyte transmigration through the vascular endothelium is a crucial step in the normal immune response. However, it is a complicated biologic process that involves many proteins and requires a coordinated effort between the leukocytes and endothelial cells (ECs). The biophysical aspects of leukocyte transmigration are also important,1 as mechanical force transmission is an essential regulator of vascular homeostasis. It is probable that the mechanical properties of the vasculature depend on both vessel size (large vessels vs microvasculature) and location (soft brain vs stiffer muscle or tumor). Further, in the cardiovascular disease of atherosclerosis, the arteries stiffen2-5 as an increased number of leukocytes penetrate the endothelium and tumor vasculature is also stiffer.6 However, it is unknown how changes in vessel stiffness affect the behavior of the ECs lining the blood vessel, or the behavior of the leukocytes migrating along and transmigrating through the endothelium. Interestingly, polymorphonuclear neutrophils are capable of sensing differences in both substrate stiffness7-9 and surface-bound adhesion proteins.8 Therefore, we would expect neutrophils to be capable of sensing similar changes that may occur in their physiologic substrate, the endothelium.
The mechanical properties of ECs are affected by a number of physiologic factors, including shear stress,10 cholesterol content,11,12 and oxidized low-density lipoprotein.13 Furthermore, neutrophil adherence to ECs increases EC stiffness, probably because of signaling cascades that induce rearrangement of the actin cytoskeleton.14,15 However, little is known about the effects of substrate stiffness on the biophysical properties of healthy or inflamed EC monolayers. Single EC stiffness increases with substrate stiffness,16 although cells in the monolayer may show different behavior than single cells, as the degree of cell-cell adhesion also contributes to cell stiffness.17
Neutrophil adherence to the endothelium has been shown to regulate EC gap formation through a cytosolic calcium-dependent mechanism.18 Myosin light chain kinase (MLCK) is activated downstream of calcium-calmodulin binding and phosphorylates myosin light chain, which activates myosin and induces EC contraction, leading to formation of gaps and subsequent regulation of neutrophil transmigration.19,20 Consistent with this cascade, leukocyte adhesion and transmigration increase the magnitude of EC traction forces exerted onto the substrate.21,22 Because cells are capable of exerting larger traction forces onto stiffer substrates,23 the MLCK-mediated signaling cascade induced by neutrophil adhesion may depend on the mechanical properties of the EC substrate, possibly leading to changes in transmigration.
In this work, we designed an in vitro model of the vascular endothelium to explore the role of EC substrate stiffness in neutrophil transmigration. Neutrophils primarily transmigrate in the microvasculature, the mechanical properties of which probably vary with health and in different regions of the body. Thus, we used fibronectin-coated polyacrylamide gel substrates of varying physiologically relevant stiffness4,24,25 (0.42-280 kPa). We plated human umbilical vein endothelial cells (HUVECs) onto the gels, allowed them to form monolayers, and activated them with TNF-α to stimulate an inflammatory response. TNF-α treatment induced significant changes in the endothelium, including softening, local alignment, enlargement, elongation, and cytoskeletal rearrangement. We then added neutrophils to the endothelium (Figure 1A) and observed transmigration. Interestingly, neutrophil transmigration increased with increasing substrate stiffness below the endothelium. To explain this, we first evaluated the effects of substrate stiffness on a range of HUVEC properties, including ICAM-1 expression, cell stiffness, F-actin organization, cell morphology, and cell-substrate adhesion. Once the HUVECs were activated with TNF-α, these properties could not account for the higher fraction of transmigrated neutrophils on stiffer substrates. Meanwhile, inhibition of MLCK or myosin II decreased transmigration on stiff substrates, whereas transmigration on soft substrates was unaffected. In addition, on stiff substrates, we observed formation of large holes in the monolayers as ECs retracted; hole formation initiated as neutrophil transmigration reached a maximum. These results provide strong evidence that neutrophil transmigration is regulated by MLCK-mediated EC contraction and that this phenomenon depends on substrate stiffness. These results may also be associated with cardiovascular disease biology, where increased arterial stiffness is coupled with increased leukocyte transmigration.
Methods
Substrate preparation and characterization
Thin polyacrylamide gels were attached to glass coverslips by a method first described by Wang and Pelham26 and used in our previous publications.8,27 Concentrations in this work included 15% acrylamide + 1.2% bis (280 kPa), 8% acrylamide + 0.2% bis (13 kPa), 8% acrylamide + 0.07% bis (5 kPa), 8% acrylamide + 0.04% bis (4 kPa), 5% acrylamide + 0.05% bis (3 kPa), 3% acrylamide + 0.1% bis (0.87 kPa), and 3% acrylamide + 0.05% bis (0.42 kPa). Gels (∼ 80-μm-thick) were coated with 0.1 mg/mL fibronectin (Sigma-Aldrich), also as previously described.8 Young moduli of the gels were determined using dynamic mechanical analysis and atomic force microscopy,8,27 and characterization of surface-bound fibronectin was done using immunofluorescence to ensure equal protein presentation with stiffness.8,27 For experiments on glass, coverslips (22 × 22 mm, Fisher Scientific) were coated with 0.1 mg/mL fibronectin for 2 hours at room temperature.
Cell culture and treatments
HUVECs were purchased from Lifeline Cell Technology and cultured as previously described.17 Human brain microvascular ECs (HBMECs; Applied Cell Biology Research Institute) were cultured as previously described.28 Cells (passages 2-5; 4 × 105 total) were plated onto fibronectin-coated glass coverslips or polyacrylamide gels and grown for approximately 48 hours until monolayer formation. Cells were then treated with 25 ng/mL human TNF-α (Fisher Scientific) for the final 24 hours before experiments. To further increase permeability of monolayers17,29-31 after TNF-α activation, HUVECs were treated with 10 ng/mL cytochalasin B (cytoB; Sigma-Aldrich) or with 1:100 dilution (10 μg/mL) of VE-cadherin antibody (V1514; Sigma-Aldrich) for 1 hour at 37°C just before experiments and then washed once with phosphate-buffered saline, as previously described.17 To inhibit MLCK or myosin II, HUVEC monolayers were pretreated with 15μM ML-7 (Sigma-Aldrich) or 15μM blebbistatin, respectively, for 8 minutes and then washed with phosphate-buffered saline before adding neutrophils. For some experiments, neutrophils were treated in suspension with 15μM ML-7 for 8 minutes, centrifuged, and resuspended in buffer solution. Treatments with dimethyl sulfoxide (DMSO; Fisher Scientific) or antihuman IgG (Fc specific) antibody (I9135; Sigma-Aldrich) were used for vehicle controls as appropriate.
Transmigration assays
Neutrophils were isolated from human blood as previously described in our work.8 Methods were approved by the University of Maryland Institutional Review Board. After TNF-α treatment, EC monolayers were washed once with phosphate-buffered saline and replaced with fresh media, or treated as described in “Cell culture and treatments.” A total of approximately 10 × 105, 5 × 105, or 2 × 105 neutrophils were plated onto the EC monolayer and allowed to gravitate down to the monolayer for approximately 30 to 60 seconds. Phase-contrast images were then captured for 30 minutes as the neutrophils migrated along and transmigrated through the EC monolayers. Microscopy for Figures 1D and 4A was completed in HUVEC culture media at 37°C, 5% CO2 and 55% humidity using an Olympus IX71 inverted microscope and a 20×/0.45 NA Ph1 objective. Images were captured with a QImaging Retiga-SRV charge-coupled device digital camera (QImaging Corporation) using IPLab Version 4.03 software. Microsoft Powerpoint was used to add shapes to the figures. Fraction of neutrophil transmigration was calculated by dividing the number of neutrophils that transmigrated in a particular 30-minute sequence by the total number of neutrophils (typically ∼ 50) in the first frame of that sequence. Neutrophils that entered or exited the field of view later during the sequence were not counted. All experiments were repeated at least 3 times. Neutrophil injury to the monolayer was quantified by counting the number of visible holes in the monolayer in images at various time points after plating neutrophils. Number of holes was normalized to total image area. Outlines of holes were traced by hand and area was quantified using ImageJ (National Institutes of Health, Bethesda, MD).
Immunostaining
HUVEC monolayers were fixed, permeabilized, and blocked for nonspecific binding as previously described.17 Cells were stained with antibodies for β-catenin (to visualize cellular borders) and vinculin (to quantify focal adhesions), or with phalloidin- tetramethylrhodamine isothiocyanate (to label F-actin) or Hoechst stain (to label cell DNA), as previously described.17 For ICAM-1 staining, nonpermeabilized HUVECs were treated with 10 μg/mL monoclonal antihuman ICAM-1 antibody (BBA3; R&D Systems) for 1 hour, followed by 5 μg/mL antimouse Alexa-488 secondary antibody (A11001; Invitrogen) for 1 hour. Fluorescence microscopy for Figure 2A was completed at room temperature on immunostained HUVECs in PBS using an Olympus IX81 inverted microscope and a 60×/1.42 NA oil objective. Images were captured with a Hamamatsu ORCA-ER charge-coupled device digital camera (Leeds Precision Instruments) using Slidebook SB 4.2.0.9 software (Intelligent Imaging Innovations). Cell morphology (area and aspect ratio) and vinculin punctate size and density were measured using the fluorescence images and ImageJ as previously described.17 Here, β-catenin-stained cell morphology was measured by applying an edge detector filter, thresholding the images, and then using the particle analyzer in ImageJ.
Atomic force microscopy
Young moduli of live HUVEC monolayers were measured using an atomic force microscope (AFM; Agilent Technologies) with a silicon nitride cantilever (Novascan) with a spherical glass SiO2 probe of diameter 5 μm. Our AFM methods have previously been described in detail.17 Gel substrates with HUVEC monolayers attached were positioned under the AFM tip, and typical force curves were captured for at least 100 different locations along each of 3 independent samples per condition. In a custom-written Matlab (The MathWorks) program, data were fit to the Hertz-Sneddon model32 for a paraboloid indenter33 :
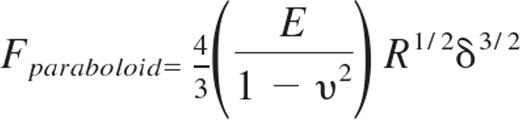
where Fparaboloid is the force exerted by the paraboloid indenter, E is the Young modulus of the cell, R is the radius of curvature of the indenter, and δ is the distance of the indenter from the sample. The cells were assumed to be nearly incompressible34 ; therefore, it was assumed that υ = 0.45 was the Poisson's ratio of the elastic half-space. The Young modulus was found for each force curve using the fitting algorithm, and average Young modulus was computed by averaging all force curves for a given condition. AFM deflection images for Figure 3A were captured in serum-free HUVEC media using the Molecular Imaging PicoScan Controller with the stage mounted on an Olympus IX71 inverted microscope, and PicoScan Version 5.3.3 software was used to control the scan.
Gels were approximately 80 μm thicker than the indentation depth (< 200nm), so that the stiffness of the glass coverslip did not affect force curves. Cells within the monolayers were probed at both the “cell body” (the raised portion) as well as at the cell “periphery” (near the base of the cell body, but not at the cell-cell junctions to avoid the thinnest part of the cell). Indentations were much smaller than the height of the sample (several microns, as measured by confocal microscopy), even at the “periphery” location. In addition, cells were probed with small forces (∼ 2 nN). Thus, it is very unlikely that the substrate stiffness below the HUVECs affected the force curves.
Statistical analysis
Statistical tests were done between pairs of data using a Student t test, or among groups of data using ANOVA, where P < .05 indicated statistical significance. After ANOVA, multiple comparisons were done using Tukey honestly significant difference criterion. All measurements reported in this article are in the format mean ± SE.
Results
Neutrophil transmigration increases with stiffness below the endothelium
We created an in vitro model of the vascular endothelium, where the blood vessel layers below the endothelium were represented by fibronectin-coated polyacrylamide gels of a range of stiffnesses (Figure 1A). HUVECs, typical cells to represent the endothelium for leukocyte transmigration in vitro, were plated at high density onto the gels to form monolayers. The endothelium was activated with TNF-α to induce an inflammatory response. Neutrophils were plated onto the endothelium, and we observed neutrophil activation and migration along the monolayer. Subsequently, a fraction of neutrophils transmigrated through the endothelium (Figure 1B-C). Neutrophil transmigration was easily observed in phase-contrast microscopy and was identified by a change in phase of the neutrophils, from white to dark (Figure 1D). Neutrophils in the process of transmigrating contained both a white portion still on top of the endothelium, as well as a dark portion already beneath the endothelium (Figure 1D white arrows). Transmigration typically occurred within about 30 seconds to several minutes. Interestingly, we observed increased neutrophil transmigration through HUVEC monolayers with increased substrate stiffness (Figure 1B; supplemental Videos 1 and 2, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). From 0.42 kPa to 5 kPa, the fraction of transmigration increased linearly with substrate stiffness (r2 = 0.99). Similar results were obtained using HBMECs (Figure 1B).
![Figure 1. An in vitro neutrophil transmigration assay was used to investigate the effects of HUVEC substrate stiffness on neutrophil transmigration. (A) HUVECs were plated onto fibronectin-coated polyacrylamide gels of various stiffnesses from 0.42 kPa to 280 kPa. After monolayer formation, HUVECs were treated with TNF-α to induce an inflammatory response. Neutrophils were isolated from human blood and plated onto the HUVEC monolayer. (B) The fraction of neutrophils that transmigrated (as described in “Transmigration assays”) was quantified as a function of the stiffness below the HUVECs. Bars represent average fraction of transmigrated cells. Error bars represent SE of 3 to 8 experiments (N = 6, 8, 4, 6, 8, 3, 7, and 3 from 0.42, 0.87, 3, 4, 5, 13, and 280 kPa, and glass [∼ 50 GPa], respectively). (C) Data from panel B, up to 5 kPa, are plotted with a linear fit (r2 = 0.99). (B-C) ***P < .001, **P < .005, or ∧P < .05, using a t test compared with 5 kPa value of same cell type. (D) Shown is an example of a phase-contrast time lapse image sequence of 4 neutrophils, 2 of which transmigrate through the endothelium. Scale bar represents 10 μm and applies to all images. T = 0 is time just before initiation of transmigration in the first cell. White arrows point to the phase-darkened portion of the neutrophil as it transmigrates through the endothelium. At T = 75 seconds, the white arrowhead points to a neutrophil that possibly initiates but does not complete transmigration. In the final frame, the 2 darkened neutrophils are between the endothelium and the gel, whereas the 2 white neutrophils are on top of the endothelium.](/view-large/figure/7219212/zh89991175350001.jpeg)
An in vitro neutrophil transmigration assay was used to investigate the effects of HUVEC substrate stiffness on neutrophil transmigration. (A) HUVECs were plated onto fibronectin-coated polyacrylamide gels of various stiffnesses from 0.42 kPa to 280 kPa. After monolayer formation, HUVECs were treated with TNF-α to induce an inflammatory response. Neutrophils were isolated from human blood and plated onto the HUVEC monolayer. (B) The fraction of neutrophils that transmigrated (as described in “Transmigration assays”) was quantified as a function of the stiffness below the HUVECs. Bars represent average fraction of transmigrated cells. Error bars represent SE of 3 to 8 experiments (N = 6, 8, 4, 6, 8, 3, 7, and 3 from 0.42, 0.87, 3, 4, 5, 13, and 280 kPa, and glass [∼ 50 GPa], respectively). (C) Data from panel B, up to 5 kPa, are plotted with a linear fit (r2 = 0.99). (B-C) ***P < .001, **P < .005, or ∧P < .05, using a t test compared with 5 kPa value of same cell type. (D) Shown is an example of a phase-contrast time lapse image sequence of 4 neutrophils, 2 of which transmigrate through the endothelium. Scale bar represents 10 μm and applies to all images. T = 0 is time just before initiation of transmigration in the first cell. White arrows point to the phase-darkened portion of the neutrophil as it transmigrates through the endothelium. At T = 75 seconds, the white arrowhead points to a neutrophil that possibly initiates but does not complete transmigration. In the final frame, the 2 darkened neutrophils are between the endothelium and the gel, whereas the 2 white neutrophils are on top of the endothelium.
An in vitro neutrophil transmigration assay was used to investigate the effects of HUVEC substrate stiffness on neutrophil transmigration. (A) HUVECs were plated onto fibronectin-coated polyacrylamide gels of various stiffnesses from 0.42 kPa to 280 kPa. After monolayer formation, HUVECs were treated with TNF-α to induce an inflammatory response. Neutrophils were isolated from human blood and plated onto the HUVEC monolayer. (B) The fraction of neutrophils that transmigrated (as described in “Transmigration assays”) was quantified as a function of the stiffness below the HUVECs. Bars represent average fraction of transmigrated cells. Error bars represent SE of 3 to 8 experiments (N = 6, 8, 4, 6, 8, 3, 7, and 3 from 0.42, 0.87, 3, 4, 5, 13, and 280 kPa, and glass [∼ 50 GPa], respectively). (C) Data from panel B, up to 5 kPa, are plotted with a linear fit (r2 = 0.99). (B-C) ***P < .001, **P < .005, or ∧P < .05, using a t test compared with 5 kPa value of same cell type. (D) Shown is an example of a phase-contrast time lapse image sequence of 4 neutrophils, 2 of which transmigrate through the endothelium. Scale bar represents 10 μm and applies to all images. T = 0 is time just before initiation of transmigration in the first cell. White arrows point to the phase-darkened portion of the neutrophil as it transmigrates through the endothelium. At T = 75 seconds, the white arrowhead points to a neutrophil that possibly initiates but does not complete transmigration. In the final frame, the 2 darkened neutrophils are between the endothelium and the gel, whereas the 2 white neutrophils are on top of the endothelium.
ICAM-1 expression does not depend on substrate stiffness
TNF-α activation of HUVECs is known to up-regulate ICAM-1, which was necessary for neutrophil transmigration according to 2 experiments that we performed: (1) If the HUVECs were not activated using TNF-α, there was low ICAM-1 expression, and we observed that only approximately 1% of neutrophils transmigrated (supplemental Figure 1); and (2) if the HUVECs were activated using TNF-α, but then ICAM-1 was blocked using an antibody, we observed the fraction of transmigration drop-off with increasing antibody concentration on all substrates (supplemental Figure 1). Thus, we quantified ICAM-1 expression on the apical surface of TNF-α–activated HUVEC monolayers using an ICAM-1 antibody and a fluorescent secondary antibody, combined with fluorescence imaging (Figure 2A). Quantification of the fluorescence intensity revealed no difference (P > .05) in ICAM-1 expression in TNF-α–activated HUVECs on 0.87-, 5-, and 280-kPa substrates (Figure 2B).

Immunostaining indicates no change in ICAM-1 expression with substrate stiffness. ICAM-1 was measured as a function of HUVEC substrate stiffness using a fluorescently tagged antibody to ICAM-1 on nonpermeabilized TNF-α-activated HUVEC monolayers. Fluorescence images were taken over many locations on the nonpermeabilized HUVEC monolayer surface (A), and intensity (in arbitrary units, au) was quantified using ImageJ software (B). Scale bar in panel A is 20 μm and applies to all images. Substrate stiffness is indicated in the upper left corner of each image in panel A. Bars represent average of at least 20 images from each of 2 independent experiments. Error bars represent SE. ANOVA indicates that P > .05 among stiffnesses.
Immunostaining indicates no change in ICAM-1 expression with substrate stiffness. ICAM-1 was measured as a function of HUVEC substrate stiffness using a fluorescently tagged antibody to ICAM-1 on nonpermeabilized TNF-α-activated HUVEC monolayers. Fluorescence images were taken over many locations on the nonpermeabilized HUVEC monolayer surface (A), and intensity (in arbitrary units, au) was quantified using ImageJ software (B). Scale bar in panel A is 20 μm and applies to all images. Substrate stiffness is indicated in the upper left corner of each image in panel A. Bars represent average of at least 20 images from each of 2 independent experiments. Error bars represent SE. ANOVA indicates that P > .05 among stiffnesses.
Stiffness of TNF-α-activated HUVEC monolayers varies little with substrate stiffness
Using AFM, we obtained deflection images (Figure 3A) and then measured the stiffness (Young modulus) of control and TNF-α-activated HUVEC monolayers on various substrates, at both the cell body and periphery locations17 (Figure 3B). Deflection images of control monolayers revealed more disorganization on soft, 0.87-kPa substrates than on stiffer 5- or 280-kPa substrates (Figure 3A top). Average stiffness of the control endothelium increased with increasing substrate stiffness at both the cell body and periphery (Figure 3B; P < .001). However, when treated with TNF-α, the HUVECs on all substrates elongated, aligned, and enlarged, and no differences were observed between the 3 substrates in the AFM deflection images (Figure 3A bottom). Further, although the stiffness of the TNF-α-activated endothelium still increased with substrate stiffness at both the cell body and periphery (Figure 3B; P < .001), the trend was much less dramatic. Interestingly, the endothelium softened with TNF-α treatment on the stiffer substrates (P < .001). In varying substrate stiffness, the stiffness of the TNF-α–activated endothelium changed only to a small degree (∼ 50 Pa at the cell body and ∼ 300 Pa at the periphery).

AFM data reveal only a slight increase in TNF-α-activated HUVEC stiffness with substrate stiffness. (A) AFM was used to obtain deflection images for HUVEC monolayers on 0.87-, 5-, and 280-kPa substrates, both under control and TNF-α-treated conditions. Deflection images are 90 μm × 90 μm. (B) AFM was also used to quantify the Young modulus (“stiffness”) of HUVEC monolayers as a function of substrate stiffness in the control (no TNF-α) and after TNF-α treatment. The stiffness of the “cell body” region (raised portion of the cell) and periphery (flattened region just around the raised portion) were quantified separately. Bars represent average stiffness from N force curves from 3 independent experiments. Error bars represent SE. N = 47, 50, and 89 on control monolayers on 0.87, 5, and 280 kPa, respectively, at the cell body. N = 381, 351, and 334 on control monolayers on 0.87, 5, and 280 kPa, respectively, at the periphery. N = 96, 121, and 160 on TNF-α-activated monolayers on 0.87, 5, and 280 kPa, respectively, at the cell body. N = 396, 357, and 399 on TNF-α-activated monolayers on 0.87, 5, and 280 kPa, respectively, at the periphery. ***P < .001 (using ANOVA). On 280 kPa, P < .001 between control and TNF-α at both cell body and periphery using Student t test.
AFM data reveal only a slight increase in TNF-α-activated HUVEC stiffness with substrate stiffness. (A) AFM was used to obtain deflection images for HUVEC monolayers on 0.87-, 5-, and 280-kPa substrates, both under control and TNF-α-treated conditions. Deflection images are 90 μm × 90 μm. (B) AFM was also used to quantify the Young modulus (“stiffness”) of HUVEC monolayers as a function of substrate stiffness in the control (no TNF-α) and after TNF-α treatment. The stiffness of the “cell body” region (raised portion of the cell) and periphery (flattened region just around the raised portion) were quantified separately. Bars represent average stiffness from N force curves from 3 independent experiments. Error bars represent SE. N = 47, 50, and 89 on control monolayers on 0.87, 5, and 280 kPa, respectively, at the cell body. N = 381, 351, and 334 on control monolayers on 0.87, 5, and 280 kPa, respectively, at the periphery. N = 96, 121, and 160 on TNF-α-activated monolayers on 0.87, 5, and 280 kPa, respectively, at the cell body. N = 396, 357, and 399 on TNF-α-activated monolayers on 0.87, 5, and 280 kPa, respectively, at the periphery. ***P < .001 (using ANOVA). On 280 kPa, P < .001 between control and TNF-α at both cell body and periphery using Student t test.
Neutrophil transmigration injures the endothelium on stiff substrates
Before neutrophil transmigration, HUVEC monolayers on soft and stiff substrates were visually intact, according to phase-contrast images. Interestingly, monolayers on soft substrates were nearly always still intact after neutrophil transmigration (Figure 4A). In contrast, neutrophil transmigration created large holes in the monolayers on stiff substrates (Figure 4B-D; supplemental Video 3), indicating EC injury. Holes initiated as the HUVECs retracted, although they retained cell-cell adhesion on the sides opposite to retraction. In some cases, significant neutrophil accumulation beneath the endothelium created large holes (supplemental Video 3) and prevented healing (Figure 4D), whereas often the endothelium was able to heal smaller holes (Figure 4E). Hole formation began at 25 minutes after plating neutrophils (about the same time when the maximum fraction of neutrophils had transmigrated), reached a peak by 45 minutes, and subsequently dropped off (Figure 5A). Hole formation was usually initiated by neutrophils, which had already transmigrated and changed in morphology from highly protrusive (supplemental Figure 2; T = 0.92 to T = 2.25 minutes), to more isotropic and spread-out (supplemental Figure 2; T = 5.83 minutes). Holes were observed similarly when 10 × 105 or 5 × 105 neutrophils were added to the endothelium, whereas very few holes were observed with 2 × 105 neutrophils (Figure 5A-B). Holes were also significantly larger on 5- or 280-kPa substrates compared with 0.87 kPa (Figure 5C).

Neutrophil transmigration on stiff substrates causes injury to the monolayer. Shown are representative phase-contrast images of the HUVEC monolayer after neutrophil transmigration on (A) a soft (0.87-kPa) substrate and (B-C) stiff (280-kPa) substrates. Time after plating neutrophils onto the HUVEC monolayers (T) is shown at the top of each image. Large holes commonly form in monolayers on stiff substrates after transmigration and are outlined in panels B and C by white dotted lines. (D) Also shown is a phase-contrast image of a monolayer on a 280-kPa substrate at approximately 2 hours after plating neutrophils. Significant neutrophil accumulation in the area of the hole has occurred. Scale bar in panel D is 50 μm and applies to images in panels A-D. (E) Shown is a time sequence of a hole forming and then healing on a 5-kPa substrate. Time after plating neutrophils is indicated at the bottom of each image. The scale bars on all images in panel E are 20 μm.
Neutrophil transmigration on stiff substrates causes injury to the monolayer. Shown are representative phase-contrast images of the HUVEC monolayer after neutrophil transmigration on (A) a soft (0.87-kPa) substrate and (B-C) stiff (280-kPa) substrates. Time after plating neutrophils onto the HUVEC monolayers (T) is shown at the top of each image. Large holes commonly form in monolayers on stiff substrates after transmigration and are outlined in panels B and C by white dotted lines. (D) Also shown is a phase-contrast image of a monolayer on a 280-kPa substrate at approximately 2 hours after plating neutrophils. Significant neutrophil accumulation in the area of the hole has occurred. Scale bar in panel D is 50 μm and applies to images in panels A-D. (E) Shown is a time sequence of a hole forming and then healing on a 5-kPa substrate. Time after plating neutrophils is indicated at the bottom of each image. The scale bars on all images in panel E are 20 μm.

EC hole formation begins when neutrophil transmigration has reached a maximum. (A) Fraction of neutrophils that have transmigrated (primary vertical axis) as a function of time after addition of neutrophils is shown for varying substrates (0.87, 5, 280 kPa). Also plotted is the number of holes per area (secondary vertical axis) as a function of time after addition of neutrophils, for varying substrates and number of neutrophils plated. (B) Data from panel A at T = 45 minutes are highlighted. Shown is the number of holes per area on each of the substrates, with varying numbers of neutrophils. (C) The area of holes at T = 45 minutes is quantified for varying substrate stiffness and number of neutrophils plated. Bars represent average. Error bars represent SE. *P < .05 (using ANOVA). ***P < .001 (using ANOVA).
EC hole formation begins when neutrophil transmigration has reached a maximum. (A) Fraction of neutrophils that have transmigrated (primary vertical axis) as a function of time after addition of neutrophils is shown for varying substrates (0.87, 5, 280 kPa). Also plotted is the number of holes per area (secondary vertical axis) as a function of time after addition of neutrophils, for varying substrates and number of neutrophils plated. (B) Data from panel A at T = 45 minutes are highlighted. Shown is the number of holes per area on each of the substrates, with varying numbers of neutrophils. (C) The area of holes at T = 45 minutes is quantified for varying substrate stiffness and number of neutrophils plated. Bars represent average. Error bars represent SE. *P < .05 (using ANOVA). ***P < .001 (using ANOVA).
Decreasing cell-cell adhesion increases transmigration on soft substrates
To determine whether endothelial cell-cell adhesion changed with substrate stiffness, we varied the degree of cell-cell adhesion by preconditioning the TNF-α–activated endothelium for one hour with one of 2 treatments: (1) 10 ng/mL cytoB or (2) VE-cadherin antibody, both of which have previously been shown to decrease cell-cell adhesion.17,29-31 With VE-cadherin antibody treatment, there was increased transmigration on soft 0.87-kPa substrates compared with the IgG antibody control (P < .05), yet no change in transmigration on stiff 280-kPa substrates compared with the control (P > .05; Figure 6A). Transmigration was not statistically different with cytoB treatment for any substrates.

MLCK mediates substrate stiffness-dependent neutrophil transmigration. (A) TNF-α–activated HUVEC monolayers were pretreated with appropriate control (DMSO or IgG antibody), cytoB, or VE-cadherin antibody (VEcad Ab) for one hour. Neutrophils were plated onto the HUVECs, and the fraction of transmigration was quantified on soft (0.87 kPa) and stiff (280 kPa) substrates. (B) TNF-α–activated HUVEC monolayers were pretreated with DMSO, blebbistatin, or ML-7. Neutrophils were plated onto the HUVECs, and the fraction of transmigration was quantified on soft (0.87 kPa), intermediate (5 kPa), and stiff (280 kPa) substrates. Also shown is the fraction of transmigration for ML-7–treated neutrophils through TNF-α–treated monolayers. (A-B) Bars represent average fraction of transmigrated cells. Error bars represent SE from at least 3 independent experiments. (A) *P < .05 with IgG antibody control using Student t test. (B) *P < .05, **P < .01 between treated monolayers and DMSO control using Student t test. (C) Schematic illustrating a possible mechanism for how pretreatment of HUVEC monolayers with ML-7 normalizes the effects of substrate stiffness in neutrophil transmigration. Before ML-7 treatment (left), neutrophil adherence to the endothelium induces a signaling cascade, which activates MLCK and results in endothelial cell contraction (black arrows) and gap formation. Because the cells can presumably exert more traction on a stiffer substrate, they are capable of creating larger gaps on the stiff substrate, ultimately allowing more neutrophils to transmigrate through. Treatment of the endothelium with ML-7 causes inhibition of contraction on the stiff substrate. The soft substrate is unaffected, possibly because contraction was already suppressed to some degree, and ML-7 treatment did not produce further effects.
MLCK mediates substrate stiffness-dependent neutrophil transmigration. (A) TNF-α–activated HUVEC monolayers were pretreated with appropriate control (DMSO or IgG antibody), cytoB, or VE-cadherin antibody (VEcad Ab) for one hour. Neutrophils were plated onto the HUVECs, and the fraction of transmigration was quantified on soft (0.87 kPa) and stiff (280 kPa) substrates. (B) TNF-α–activated HUVEC monolayers were pretreated with DMSO, blebbistatin, or ML-7. Neutrophils were plated onto the HUVECs, and the fraction of transmigration was quantified on soft (0.87 kPa), intermediate (5 kPa), and stiff (280 kPa) substrates. Also shown is the fraction of transmigration for ML-7–treated neutrophils through TNF-α–treated monolayers. (A-B) Bars represent average fraction of transmigrated cells. Error bars represent SE from at least 3 independent experiments. (A) *P < .05 with IgG antibody control using Student t test. (B) *P < .05, **P < .01 between treated monolayers and DMSO control using Student t test. (C) Schematic illustrating a possible mechanism for how pretreatment of HUVEC monolayers with ML-7 normalizes the effects of substrate stiffness in neutrophil transmigration. Before ML-7 treatment (left), neutrophil adherence to the endothelium induces a signaling cascade, which activates MLCK and results in endothelial cell contraction (black arrows) and gap formation. Because the cells can presumably exert more traction on a stiffer substrate, they are capable of creating larger gaps on the stiff substrate, ultimately allowing more neutrophils to transmigrate through. Treatment of the endothelium with ML-7 causes inhibition of contraction on the stiff substrate. The soft substrate is unaffected, possibly because contraction was already suppressed to some degree, and ML-7 treatment did not produce further effects.
Inhibition of MLCK normalizes effects of substrate stiffness
We investigated the role of MLCK-mediated EC contraction in neutrophil transmigration as a function of substrate stiffness using the selective MLCK inhibitor ML-735 (Figure 6B). Transmigration through ML-7–treated HUVECs on stiff substrates (5 kPa and 280 kPa; 58% ± 6% and 67% ± 5% transmigration, respectively) was reduced nearly to the level as on soft substrates with the same treatment (0.87 kPa; 45% ± 5% transmigration; Figure 6B). On 5 kPa and 280 kPa, transmigration was significantly reduced with respect to the DMSO vehicle control (Figure 6B; P < .01), whereas transmigration on the soft 0.87-kPa substrate was not affected. Treatment of HUVECs with blebbistatin to inhibit myosin II had the same effect as ML-7 treatment (Figure 6B). Very few holes formed on all substrates with ML-7 treatment. ICAM-1 expression was not affected by ML-7 treatment on any substrate (supplemental Figure 3). As expected, transmigration of ML-7-treated neutrophils through untreated (TNF-α only) ECs was reduced on all substrates (Figure 6B).
Discussion
Neutrophil transmigration is an important physiologic process in the normal immune response and occurs in blood vessels whose mechanical properties probably depend on location within the body, size, and health. In this work, we evaluated the effects of vessel stiffness on both the endothelium properties and neutrophil transmigration. In our in vitro model, we observed that the fraction of transmigrating neutrophils increased with increasing stiffness below the endothelium, in both large-vessel endothelium (HUVECs) and also in microvasculature endothelium (HBMECs) (Figure 1B). Because substrate stiffness is known to control many biologic and biomechanical properties of cells, we evaluated an array of properties of the endothelium to explain this behavior, including EC monolayer ICAM-1 expression, stiffness, morphology, cell-substrate adhesion, cell-cell adhesion, and cell contraction. Our results provide strong evidence that substrate stiffness changes cell-cell adhesion through MLCK-dependent cell contraction.
Previous work regarding neutrophil transmigration has mostly used glass (stiffness ∼ 50 GPa) or transwell filters as substrates. It is probable that blood vessel stiffness depends not only on the size and health of the vessel but also on the location within the body because tissues, such as brain, muscle, and tumors, vary in mechanical properties (see further discussion below).36,37 Therefore, it is more physiologically relevant to perform in vitro transmigration experiments on softer substrates in the kilopascal range of stiffness. Here, we vary the substrate stiffness of the endothelium in a controlled way to evaluate its contribution to neutrophil transmigration.
Our results indicate that neutrophil transmigration involves an interplay between EC contraction, neutrophil contraction, and ICAM-1 signaling. It is known that neutrophil binding to ICAM-1 on the surface of the endothelium18-20,38 initiates MLCK-dependent EC contraction, which should depend on the stiffness below the endothelium because cells are able to exert larger traction forces on stiffer substrates.23 Here, inhibition of myosin II-dependent EC contraction by targeting myosin II or upstream MLCK in HUVECs not only reduced transmigration on intermediate (5 kPa) and stiff (280 kPa) substrates compared with the vehicle control but reduced it nearly to the level of transmigration on soft substrates (Figure 6B). Meanwhile, transmigration on the soft (0.87 kPa) substrate was unaffected (Figure 6B), probably because contraction was already suppressed because of the ECs' inability to exert large traction forces on a soft substrate.23 Stiff substrates probably promote contraction through mechanotransduction events in EC integrin-to-extracellular matrix adhesions, possibly through talin activation.39 Consistent with these results, it has been shown that inhibition of contraction reduces cell stiffness.40
Together, these results support the situation described by the schematic in Figure 6C. Contractile forces (black arrows) resulting from actin-myosin activation are larger on stiff substrates and create intercellular gaps that are more permissible for neutrophil penetration; contraction is suppressed by soft substrates. When MLCK is inhibited, contractile forces on stiff substrates are reduced, leading to less transmigration, whereas inhibition of MLCK does not further reduce contractile forces on soft substrates, leading to no change in transmigration. The schematic in Figure 7 summarizes how our results fit into the known MLCK-mediated signaling pathway initiated by neutrophil adhesion to the endothelium.

Signaling cascade initiated by neutrophil adhesion to ECs is affected by substrate mechanical properties. This flow chart indicates how our results fit into the signaling cascade initiated by neutrophil adhesion to ICAM-1 on the endothelium. **Cellular components were measured using various microscopic techniques. Components outlined with dotted lines were varied experimentally.
Signaling cascade initiated by neutrophil adhesion to ECs is affected by substrate mechanical properties. This flow chart indicates how our results fit into the signaling cascade initiated by neutrophil adhesion to ICAM-1 on the endothelium. **Cellular components were measured using various microscopic techniques. Components outlined with dotted lines were varied experimentally.
In addition to EC contraction, neutrophil contraction also contributes to transmigration, as inhibition of MLCK in neutrophils reduced (but did not block) transmigration on all substrates, including the soft ones. The result that transmigration was not completely blocked on soft substrates suggests that ICAM-1 levels (and probably other signaling molecules, such as platelet EC adhesion molecule-1) were high enough to support transmigration, even though neutrophil contraction was inhibited by drug treatment and EC contraction was reduced by substrate stiffness. Indeed, ICAM-1 is an important player and is necessary for transmigration, as the fraction of transmigrated neutrophils was directly related to the amount of ICAM-1 on the surface of the endothelium, even on stiff substrates where contraction was higher (supplemental Figure 1). Specifically, when the ECs were not treated with TNF-α (low ICAM-1), we observed only approximately 1% transmigration on all substrates (supplemental Figure 1). These results provide evidence that, even though EC contraction promotes transmigration, it alone is not sufficient to support transmigration; signaling by adhesion molecules such as ICAM-1 is also necessary.
TNF-α is known to increase EC permeability to macromolecules both in vivo and in vitro41 through long-term reorganization of junctional proteins, such as occludin, junctional adhesion molecule-A, and the membrane-associated protein ZO-1, and not via Rho-, ROCK-, and MLCK-mediated EC contraction.42 Specifically, inhibiting MLCK during TNF-α treatment does not interfere with the increase in EC permeability over the 24-hour activation period.42 Therefore, in our experiments, treatment of the HUVECs with ML-7 for one hour before transmigration assays did not reverse the effects of TNF-α–mediated permeability. Thus, the decrease in neutrophil transmigration that we observed through ML-7–treated endothelium on intermediate (5 kPa) and stiff (280 kPa) substrates (Figure 6B) reflects changes in EC contraction because of substrate stiffness (Figures 6C, 7) and not because of a reversal of TNF-α–mediated permeability.
In explaining the result that stiff substrates promote neutrophil transmigration, we must also rule out possible effects of substrate stiffness on other properties of the endothelium. We have previously demonstrated that neutrophils are sensitive both to the stiffness of their substrate and also to the concentration of adhesion protein on the surface of their substrate.8 In our in vitro model, before transmigration, the endothelium is the neutrophil “substrate.” Therefore, it makes sense that neutrophils would be sensitive to changes in either the amount of adhesion protein on the apical surface of the HUVECs, or to the actual stiffness of the monolayer. However, because ICAM-1 immunostaining indicated no change in the amount of ICAM-1 with substrate stiffness (Figure 2), it is unlikely that differences in this adhesion protein can explain the differences in transmigration behavior. Further, treatment with TNF-α to induce inflammatory conditions softened the monolayers on stiff substrates (Figure 3B), which is in agreement with previous studies on glass.43 After this treatment, the average stiffness of the endothelium varied only 50 to 300 Pa between substrates (Figure 3B), depending on location. Although the monolayer is fairly heterogeneous,17 it is unlikely that neutrophil mechanosensing of endothelium stiffness caused an increase in transmigration from 51%-91% from 0.42 kPa to 280 kPa, as mechanosensing typically occurs with changes in stiffness in the kilopascal range, not hundreds of Pascals.8 However, it is still possible that the small but significant increase in EC stiffness is a reflection of increased contractility on stiffer substrates. In addition to ICAM-1 expression and cell stiffness, we also investigated whether HUVEC F-actin organization, morphology (area and aspect ratio), or focal adhesions (number per area or size) could explain the increased transmigration on stiffer substrates; however, none of these properties could account for the behavior (supplemental Figures 4-7; supplemental Data). Finally, neutrophils adhered to and migrated on endothelium on all substrates, indicating that their ability to move to a suitable location for transmigration was not hindered on soft substrates (supplemental Videos 1 and 2).
The large holes in the endothelium that we observed as a result of neutrophil transmigration on stiff substrates (Figure 4B-D; supplemental Video 3) suggest 3 possibilities: (1) there was less EC-substrate adhesion on stiff substrates; (2) there was less endothelial cell-cell adhesion on stiff substrates; and (3) increased transmigration resulted in elevated neutrophil protease activity, causing cleavage of cell-substrate and cell-cell adhesions on stiff substrates. Because initial focal adhesion (FA) patterns were the same with substrate stiffness, as measured by vinculin punctate staining (supplemental Figure 7), we ruled out the first explanation. However, it could still be possible that neutrophil adhesion and subsequent transmigration caused rearrangement of the endothelium FA structure as a function of substrate stiffness. Evidence for this arises from supplemental Figure 2, where a change in neutrophil morphology, transitioning from protrusive to round, probably disrupted EC-substrate adhesions. This would be an interesting avenue for future exploration. Second, a reduction in cell-cell adhesion on stiff substrates may partially explain hole formation. The result that neutrophil transmigration was increased on soft substrates, but not stiff substrates, by decreasing cell-cell adhesion with a VE-cadherin antibody (Figure 6A) suggests that cell-cell adhesion was compromised on stiff substrates to a greater degree than soft substrates after TNF-α treatment; this could promote hole formation as well as neutrophil transmigration.
A third explanation for the formation of holes in the monolayers could be the result of protease release by the neutrophils as they migrated between the gels and the endothelium. Neutrophil injury to the endothelium has previously been observed in vitro44,45 and can be inhibited by tryptic, elastase, and serine protease inhibitors.45 In this case, hole formation on the stiff substrates could be because of the fact that nearly twice as many neutrophils transmigrated compared with soft substrates (Figure 1B), resulting in more proteases. To address this, we simply halved the number of neutrophils added to the surface of the endothelium (10 × 105 to 5 × 105) and observed transmigration (Figure 5A). Interestingly, holes still formed with 5 × 105 neutrophils and were similar in density and size to 10 × 105 neutrophils (Figure 5B-C), indicating that it was not simply the number of transmigrated neutrophils that influenced hole formation on the stiff substrates. Thus, it seems plausible that increased contractile forces in ECs on stiff substrates promote retraction as neutrophil proteases act on the ECs. Further evidence for this hypothesis is our observation that ML-7 inhibited hole formation in ECs on stiff substrates.
Further reduction of the number of neutrophils plated (2 × 105) eliminated hole formation, signifying that neutrophils were responsible for hole formation and that a threshold number was needed. Interestingly, the kinetics of hole formation aligned with neutrophil transmigration; that is, hole formation began approximately 25 minutes after plating neutrophils, a few minutes after the maximum fraction of neutrophils had transmigrated (Figure 5A). Thus, it is probable that protease release by neutrophils, in combination with increased contractile forces to promote retraction in ECs on stiffer substrates, led to significant EC injury.
In this work, we varied the stiffness of the EC substrate to mimic changes that occur during cardiovascular disease5 or cancer,6 and possibly throughout the body depending on tissue location. Clinically, pulse-wave velocity measurements have been used to determine that diseased arteries are consistently stiffer in patients with atherosclerosis or hypertension versus healthy patients.2,3 Further, in vivo AFM measurements of aortic vessels in living rats show a significant increase in blood vessel stiffness with vasodilation and softening with vasoconstriction.46 It is thought that the microcirculation also undergoes stiffening during hypertension, as pulse-wave velocity waves reflect deep into the microvasculature.47 Thus, it is clear that cardiovascular disease leads to blood vessel stiffening, although further work has been necessary to demonstrate that the subendothelial matrix specifically varies in stiffness with disease.
AFM has been used to quantify ex vivo blood vessel mechanical properties. For example, physiologic porcine aorta stiffness is 5-8 kPa.24 Further, the arteries of ApoE-null mice, a model of atherosclerosis, are stiffer than wild-type mice; healthy arteries measure 5 kPa, whereas ApoE knockout vessels measure 28 kPa.4 These measurements were made after the endothelium was scraped away and thus represent the stiffness of the EC substrate in an actual artery. Interestingly, the subendothelial and endothelial layers of bovine carotid arteries are similar in stiffness, approximately 2.5 kPa.25 In another study, injury to the femoral artery increased vessel stiffness from 3 kPa to 10 kPa, indicating that injury can also affect stiffness of the vasculature.48 Here, we observed a linear increase in transmigration from very soft (0.42-0.87 kPa) to the “healthy” range of subendothelial layer stiffness (3-5 kPa), reaching a threshold value in the “disease” stiffness range, from 13 or 280 kPa (Figure 1B-C).
It is also possible that the stiffness of blood vessels, specifically the microvasculature where neutrophils most often transmigrate, depends on the mechanical properties of the surrounding tissue. For example, the stiffness of brain (0.3-0.5 kPa) is much less than that of collagenous bone (∼ 100 kPa).27,37 Further, vasculature within the core of a tumor is stiffer than the surrounding vasculature.6 Thus, “healthy stiffness” probably depends on location within the body as well as size of vessel, and our substrates span a large range of physiologic stiffnesses, from 0.42 kPa to 280 kPa. Our lower range of subendothelial matrix stiffness (0.42-0.87 kPa) could be relevant in brain microvasculature where the blood vessel microenvironment is very soft (0.3-0.5 kPa as discussed earlier in this paragraph) or in development of future cardiovascular disease-targeting drugs, which return elasticity to blood vessels; it would be important to understand how these potential drugs affect EC biomechanics and the immune response.
In conclusion, we have developed an in vitro model of the vascular endothelium using polyacrylamide gels of various stiffnesses to investigate the effects of vasculature stiffness, both on the endothelium and on neutrophil transmigration. Interestingly, we observed increased neutrophil transmigration through ECs on stiffer substrates. Our results provide strong evidence that neutrophil transmigration is regulated by MLCK-mediated EC contraction and that substrate stiffness mediates this response. Neutrophil transmigration also promotes EC retraction and large hole formation in endothelium on stiff substrates, an event that is further indicative of increased contractility. Myosin light chains may also be phosphorylated by Rho kinase during neutrophil transmigration49 (Figure 7); therefore, further experiments could test whether Rho kinase-dependent cell contraction also plays a role in how the endothelium substrate stiffness affects neutrophil transmigration. In addition, it would be interesting to investigate how MLCK-mediated EC contraction regulates paracellular versus transcellular neutrophil transmigration. Our results suggest that neutrophil transmigration may vary with blood vessel mechanical properties, depending on location within the body and size. Further, these results may be associated with cardiovascular disease biology, where increased arterial stiffness is coupled with increased leukocyte transmigration. In addition, this work may be relevant to cancer cell metastasis or stem cell homing, both of which involve cell transmigration across the endothelium.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Drs Peter Davies, Martin Schwartz, and Silvia Muro for insightful conversations and the laboratory of Dr Silvia Muro for generously providing HBMECs.
This work was supported by a National Science Foundation (Graduate Research Fellowship, K.M.S.), National Institutes of Health (National Research Service Award F31NS068028, K.M.S.), and National Science Foundation (award CMMI-0643783, H.A.-E.).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Neurologic Disorders and Stroke or the National Institutes of Health.
National Institutes of Health
Authorship
Contribution: K.M.S. designed and performed experiments, analyzed and interpreted the data, and wrote the manuscript; and H.A.-E. contributed to research design, interpreted the data, and reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Helim Aranda-Espinoza, 3138 Jeong H. Kim Engineering Bldg, Fischell Department of Bioengineering, University of Maryland, College Park, MD 20742; e-mail: helim@umd.edu.

