

Abstract
Pyruvate kinase (PK) deficiency is a congenital hemolytic anemia with wide-ranging clinical symptoms and complications associated with significant morbidity and reduced health-related quality of life in both children and adults. The management of patients with PK deficiency has been historically challenging due to difficulties in the diagnostic evaluation, heterogeneity of clinical manifestations, and treatment options limited to supportive care with transfusions and splenectomy. An oral allosteric PK activator, mitapivat, is now a clinically available disease-modifying treatment for adults with PK deficiency. Phase 2 and 3 clinical trials of mitapivat have demonstrated sustained improvements in hemolytic anemia, hematopoiesis, and quality of life in many adults with PK deficiency and a generally reassuring safety profile with continued dosing. Additional long-term benefits include rapid and ongoing reduction in iron overload and potential stabilization of bone health. Clinical trials of treatment with mitapivat in children with PK deficiency are ongoing. In addition to disease-modifying treatment with PK activators, gene therapy is a potentially curative treatment currently under evaluation in clinical trials. With the availability of disease-targeted therapies, accurately diagnosing PK deficiency in patients with chronic hemolytic anemia is critical. PK activation and gene therapy have the potential to change the natural history of PK deficiency by improving clinical manifestations and patient quality of life and decreasing the risk of long-term complications.
Learning Objectives
Evaluate the available data for pyruvate kinase (PK) activators for the treatment of PK deficiency
Apply concepts to generate an approach to the diagnostic evaluation and treatment of PK deficiency
CLINICAL CASE
A 32-year-old man presents for a new consultation for chronic hemolytic anemia. He was evaluated for anemia in early childhood and told that he had hereditary spherocytosis. He has not seen a hematologist in many years. He received several red cell transfusions in the first few years of his life but has not since been transfused. At age 10 years, he had cholecystitis and underwent laparoscopic cholecystectomy. At the consult visit, he reports longstanding fatigue that has worsened over the prior 5 years. He works full-time as teacher but is unable to exercise, and he limits activities with his children because of fatigue and shortness of breath. He would like to know how he can improve his energy.
Introduction
Pyruvate kinase (PK) deficiency is rare congenital hemolytic anemia associated with a wide spectrum of symptoms and complications due to chronic hemolysis. PK is a tetrameric glycolytic enzyme that catalyzes the conversion of phosphoenolpyruvate to pyruvate and leads to adenosine triphosphate (ATP) production. PK deficiency is an autosomal recessive condition caused by mutations in the PKLR gene, which encodes the red cell and liver specific isoforms PK-R and PK-L. Given the reliance of mature red cells on glycolysis as their primary energy source, PKLR mutations cause an insufficiency in ATP, which leads to red cell dehydration and membrane abnormalities that cause chronic hemolysis and ineffective erythropoiesis.1-4 PK-deficient reticulocytes are particularly susceptible to injury in the hypoxic spleen due to high ATP needs and reliance on glycolysis rather than oxidative phosphorylation in the splenic environment.5 The glycolytic intermediate 2,3-biphosphoglycerate (2,3-BPG) is increased in PK deficiency, causing a shift in the hemoglobin-oxygenation dissociation curve with a consequent increase in tissue oxygenation; in turn, greater tissue oxygenation can lead to improved tolerance of anemia in some patients.6
More than 350 pathogenic PKLR mutations have been reported, and most patients have compound heterozygous PKLR variants, leading to wide genotypic variability. The majority have at least one missense PKLR mutation encoding single amino acid changes that affect PK catalytic activity, stability, or expression. Prior studies have not shown a strong genotype-phenotype relationship, including among siblings, although assessment has been limited by small patient cohorts and extensive combinations of genotypes.7
The estimated prevalence ranges from 1 to 8 in 1 000 000.8,9 Diagnostic testing should be considered in patients of all ages with chronic nonimmune hemolysis and requires high clinical suspicion given the wide-ranging clinical phenotype and nonspecific red cell morphology (Figures 1 and 2). When available, both evaluation of red cell enzyme activity, with a low PK:hexokinase ratio, and PKLR genetic testing should be pursued to allow the diagnosis to be made based on the combination of test results, since each has limitations; expert guidelines for diagnostic evaluation are available.10,11 Although useful for diagnosis, PK enzyme activity does not correlate with clinical severity and is not predictive of clinical course.11
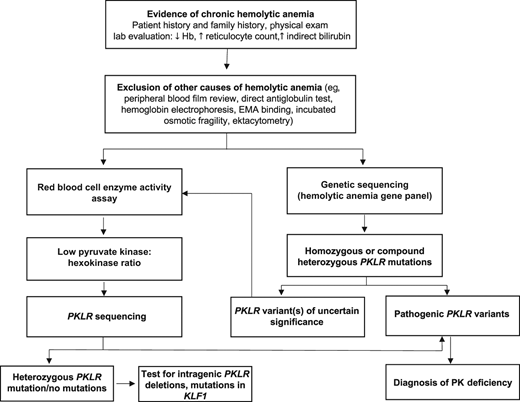
Algorithm for the diagnostic evaluation of PK deficiency. An accurate diagnosis of PK deficiency is key for managing patients with disease-targeted treatments. Diagnostic testing should be considered in patients of all ages with chronic hemolytic anemia. After hemolysis has been established (low hemoglobin, increased reticulocyte count and indirect bilirubin level), autoimmune hemolytic anemia, hemoglobinopathies, and membranopathies should be excluded. The peripheral blood film in PK deficiency often does not have specific red cell morphology except polychromasia. Once the more common causes of hemolysis are excluded, a red cell enzyme panel or a hemolytic anemia gene panel may be pursued. Falsely normal PK enzyme activity levels may occur with recent red cell transfusions, reticulocytosis, and/or sample contamination with other blood cells. PK enzyme activity is red cell age dependent and will be low in comparison to other red cell age dependent enzymes, such as hexokinase. In those with a low PK:hexokinase ratio on enzyme testing, PKLR genetic testing should be pursued given that low PK activity can also be found in carriers of PK deficiency and in those with mutations in KLF1.38 Up to 20% of patients currently tested will be found to have a PKLR variant of uncertain significance.12 In these patients, a low PK:hexokinase ratio can confirm the diagnosis. EMA, eosin-5’-maleimide.
Algorithm for the diagnostic evaluation of PK deficiency. An accurate diagnosis of PK deficiency is key for managing patients with disease-targeted treatments. Diagnostic testing should be considered in patients of all ages with chronic hemolytic anemia. After hemolysis has been established (low hemoglobin, increased reticulocyte count and indirect bilirubin level), autoimmune hemolytic anemia, hemoglobinopathies, and membranopathies should be excluded. The peripheral blood film in PK deficiency often does not have specific red cell morphology except polychromasia. Once the more common causes of hemolysis are excluded, a red cell enzyme panel or a hemolytic anemia gene panel may be pursued. Falsely normal PK enzyme activity levels may occur with recent red cell transfusions, reticulocytosis, and/or sample contamination with other blood cells. PK enzyme activity is red cell age dependent and will be low in comparison to other red cell age dependent enzymes, such as hexokinase. In those with a low PK:hexokinase ratio on enzyme testing, PKLR genetic testing should be pursued given that low PK activity can also be found in carriers of PK deficiency and in those with mutations in KLF1.38 Up to 20% of patients currently tested will be found to have a PKLR variant of uncertain significance.12 In these patients, a low PK:hexokinase ratio can confirm the diagnosis. EMA, eosin-5’-maleimide.
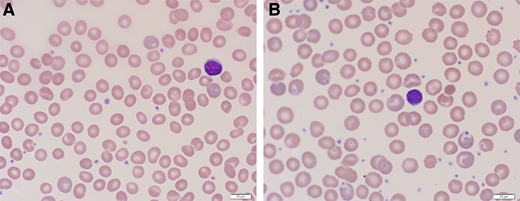
Peripheral blood film from a patient with PK deficiency with an intact spleen (A) and after splenectomy (B). The red cell findings in PK deficiency with an intact spleen are mild. Nonspecific or bland red cell morphology in a patient with a congenital hemolytic anemia should raise suspicion for an enzymopathy. After splenectomy, the peripheral blood film is notable for moderate polychromasia and echinocytes.
Peripheral blood film from a patient with PK deficiency with an intact spleen (A) and after splenectomy (B). The red cell findings in PK deficiency with an intact spleen are mild. Nonspecific or bland red cell morphology in a patient with a congenital hemolytic anemia should raise suspicion for an enzymopathy. After splenectomy, the peripheral blood film is notable for moderate polychromasia and echinocytes.
CLINICAL CASE (continued)
The patient has a hemoglobin (Hb) level of 8.2 g/dL, reticulocyte count 20%, indirect bilirubin 3 mg/dl, ferritin 1100 ng/mL, and a peripheral blood film with polychromasia but otherwise bland red cell morphology without spherocytes (Figure 2). His direct antiglobulin test is negative, and a hemoglobin electrophoresis is normal. His PK activity is 1.4 EU/g Hb (normal range: 3.2-6.5 EU/g Hb) with a hexokinase (HK) activity of 1.07 EU/g Hb (normal range: 0.14-0.37 EU/g Hb) and PK:HK ratio of 1.31 (reference normal PK:HK ratio range: 8.7-22.5). A hemolytic anemia gene panel reveals two pathogenic missense PKLR variants (1529G>A, 1456C>T) consistent with a diagnosis of PK deficiency. Liver iron quantification with magnetic resonance imaging (MRI) is 7 mg/g dry weight liver. A dual energy x-ray absorptiometry (DXA) scan shows the lowest bone mineral density Z-score is −2.5 at the femoral neck, consistent with osteoporosis.
Symptoms and complications
PK deficiency has a wide clinical spectrum due to both chronic hemolysis and complications of supportive treatment (Figure 3). Severe manifestations can occur in utero that result in hydrops fetalis, growth restriction, and/or prematurity.12 Newborns may have a presentation ranging widely from compensated anemia to neonatal hyperbilirubinemia to critical illness with organomegaly, hyperferritinemia, and/or liver failure.12,13 Infants and children may have poor growth and irritability related to anemia. Affected individuals of all ages may have jaundice, scleral icterus, fatigue, cognitive effects, or limited exercise tolerance, all of which can affect quality of life.14 Clinical findings include splenomegaly, iron overload (transfused and nontransfused), osteopenia and osteoporosis, gallbladder disease, extramedullary hematopoiesis, lower extremity ulceration, and pulmonary hypertension. Iron overload, both due to transfusions and increased iron absorption, can cause significant morbidity, including cardiac failure, liver disease, and endocrine dysfunction.12,15,16 Regular monitoring of ferritin and MRI guides treatment with iron chelation.17 Exacerbations of hemolytic anemia may occur with infections and pregnancy.12 With advancing age, symptoms associated with anemia may increase in the presence of additional comorbidities.
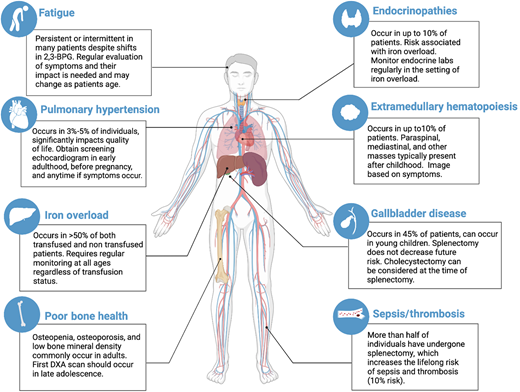
Non–PK activator treatment strategies and associated complications
Prior to the approval of PK activators, management of PK deficiency included supportive treatments of red cell transfusions and splenectomy and potentially curative treatment with allogeneic hematopoietic stem cell transplant (HSCT). Early results from a gene therapy trial are promising. Decision-making about treatment with a PK activator should be evaluated in the context of these management options.
Red cell transfusion therapy is initiated based on growth in children, symptoms of anemia with impact on quality of life, complications, and, to some extent, baseline hemoglobin. The requirement for transfusions decreases over childhood, likely due to a decrease in the frequency of viral infections and the historic timing of splenectomy in children.12 Erythropoiesis is supported with folic acid, particularly in the setting of dietary limitations or pregnancy. Symptoms of chronic anemia may increase during adulthood, leading to the initiation of regular transfusions.
Splenectomy is often considered in childhood for patients who undergo transfusions or have a poor quality of life due to anemia.18 After splenectomy, many, but not all, patients have an improvement in hemoglobin (mean hemoglobin increase 1.5 g/dL) and transfusion burden.19 Splenectomy leads to a paradoxical increase in the reticulocyte count. Those with more severe hemolysis are less likely to benefit from splenectomy.12,14 Hemolysis continues after splenectomy with an ongoing risk of associated complications in addition to the lifelong risk of sepsis and thrombosis.12
Although HSCT is a curative treatment, there are no clear indications for transplant for PK deficiency. Published cohort studies reveal a higher rate of morbidity and mortality associated with transplant when compared with supportive care.19,20 A global cohort study described 16 patients with PK deficiency who underwent transplantation in Europe and Asia (median age 6.5 years, median follow up 2.3 years).21 Outcomes were poor, with grade 3-4 graft-versus-host disease in 7 of 16 patients and a 3-year cumulative survival of 65%.
Gene therapy also represents a potential curative option.22-24 A phase 1 clinical trial evaluated the safety of autologous gene therapy for PK deficiency using myeloablative conditioning with lentivirus-based genetically modified hematopoietic stem cells with the corrected PKLR gene. Interim trial results of 2 adults have demonstrated a post–gene therapy normal hemoglobin, improvement in both hemolytic markers and quality of life, and a continuous transfusion-free period with 24 months of follow-up. To date, there have been no serious adverse events related to the product.25 Long-term safety and efficacy data collection from the phase 1 trial is ongoing; a phase 2 trial is planned.
PK activators for treatment of PK deficiency
Several oral PK activators are in clinical trials for treatment of red cell disorders. Mitapivat (AG-348, Agios Pharmaceuticals) is an oral allosteric erythrocyte PK activator and is the only PK activator that has been evaluated for treatment of PK deficiency (Table 1). Mitapivat binds at the dimer-dimer interface at a separate site from fructose 1,6-bisphosphate on the PK-R tetramer. The drug is orally bioavailable with or without food, with a steady state reached after 1 week with twice-daily dosing. Mitapivat is cleared through hepatic metabolism by cytochrome P450 enzymes and has an off-target effect of mild dose-dependent reversible inhibition of aromatase.26 Ex vivo human studies demonstrate increased PK enzyme activity of both wild-type and variant PK and improvement in thermostability in variant PK with exposure to mitapivat.27,28 In samples from patients with PK deficiency, incubation with mitapivat improved red cell deformability as measured by osmotic gradient ektacytometry, improved proliferation of erythroid progenitor cells, and led to an increase in ATP in a dose dependent manner.28
Clinical trial data of PK activators for treatment of PK deficiency
| Clinical trial | Study information | Summary of main findings | Authors, reference |
|---|---|---|---|
| Healthy Volunteer Trial of Mitapivat | Phase 1, healthy volunteers | • One grade 3 TEAE in multiple ascending dose study at a dose of 700 mg every 12 hours (abnormal liver function tests). Hormone changes consistent with reversible, dose-dependent aromatase inhibition. • Pharmacodynamic profile: maximal decrease 2,3-BPG 24 hours postdose; maximal increase in ATP between 8-14 days of dosing, durable for 48-72 h after dosing supporting twice daily dosing. | Yang H, Merica E, Chen Y, et al.26 |
| Healthy Volunteer Trial of Etavopivat | Phase 1, healthy volunteers | • Treatment emergent events were mild to moderate; none led to discontinuation. No changes in hormone levels. • Pharmacodynamic profile: maximal decrease 2,3-BPG 24 hours postdose; maximal increase in ATP by day 8 of dosing, durable for 120 h after dosing supporting once daily dosing | Forsyth S, Schroeder P, Geib J, et al.37 |
| DRIVE-PK trial (mitapivat) | Phase 2, adults with PK deficiency, not regularly transfused (<4 transfusions in prior 12 months) | • 26/52 (50%) of adults had an increase in Hb ≥1 g/dL; mean maximum Hb increase 3.4 g/dL • Increase in Hb occurred in median of 10 days. • Genotype-hemoglobin response relationship with all Hb responders with at least one missense PKLR mutation. • Well-tolerated, most common adverse events were headaches, insomnia, nausea, generally resolving within 1 week. Rebound hemolysis seen with abrupt discontinuation of drug. | Grace RF, Rose C, Layton MD, et al.29 |
| ACTIVATE trial (mitapivat) | Phase 3, adults with PK deficiency, not regularly transfused (<4 transfusions in prior 12 months) and at least one missense PKLR mutation | • 16/40 (40%) receiving mitapivat met the primary endpoint (Hb ≥1.5 g/dL, at least 2 timepoints) compared with 0/40 (0%) receiving placebo • Hemoglobin increase correlated with improvements in markers of hemolysis and hematopoiesis and disease-specific patient reported outcome measures. • Well-tolerated; adverse events (most common: nausea, headache) were similar in the placebo and mitapivat arms. | Al-Samkari H, Galactéros F, Glenthøj A, et al.30 |
| ACTIVATE-T trial (mitapivat) | Phase 3, adults with PK deficiency receiving ≥6 transfusions in prior 12 months and at least 1 missense PKLR mutation | • 10/27 (37%) of adults receiving mitapivat with ≥33% reduction in transfusion burden. • 6/27 (22%) transfusion free after starting mitapivat. • Adverse events included headache, nausea, increase in liver enzymes. | Glenthøj A, van Beers EJ, Al-Samkari H, et al.31 |
| ACTIVATE/ACTIVATE-T open label long-term extension study (mitapivat) | Extension study of ACTIVATE/ACTIVATE-T trials | • ACTIVATE: 39.5% (15/38) randomized to placebo had a Hb response after switching to mitapivat; response by hemoglobin, hemolytic markers, and patient-reported outcomes has been sustained for more than 3 years in the majority. • ACTIVATE-T: 37% (10/27) have met criteria for transfusion response for more than 3 years; 6 patients have remained transfusion free for more than 3 years. • Well-tolerated, no new safety findings. • Decrease in iron overload by liver iron concentration and other iron markers seen in both studies with ongoing improvements over time. • Sustained stability in bone mineral density by DXA scan. | Multiple references32-36 |
| Clinical trial | Study information | Summary of main findings | Authors, reference |
|---|---|---|---|
| Healthy Volunteer Trial of Mitapivat | Phase 1, healthy volunteers | • One grade 3 TEAE in multiple ascending dose study at a dose of 700 mg every 12 hours (abnormal liver function tests). Hormone changes consistent with reversible, dose-dependent aromatase inhibition. • Pharmacodynamic profile: maximal decrease 2,3-BPG 24 hours postdose; maximal increase in ATP between 8-14 days of dosing, durable for 48-72 h after dosing supporting twice daily dosing. | Yang H, Merica E, Chen Y, et al.26 |
| Healthy Volunteer Trial of Etavopivat | Phase 1, healthy volunteers | • Treatment emergent events were mild to moderate; none led to discontinuation. No changes in hormone levels. • Pharmacodynamic profile: maximal decrease 2,3-BPG 24 hours postdose; maximal increase in ATP by day 8 of dosing, durable for 120 h after dosing supporting once daily dosing | Forsyth S, Schroeder P, Geib J, et al.37 |
| DRIVE-PK trial (mitapivat) | Phase 2, adults with PK deficiency, not regularly transfused (<4 transfusions in prior 12 months) | • 26/52 (50%) of adults had an increase in Hb ≥1 g/dL; mean maximum Hb increase 3.4 g/dL • Increase in Hb occurred in median of 10 days. • Genotype-hemoglobin response relationship with all Hb responders with at least one missense PKLR mutation. • Well-tolerated, most common adverse events were headaches, insomnia, nausea, generally resolving within 1 week. Rebound hemolysis seen with abrupt discontinuation of drug. | Grace RF, Rose C, Layton MD, et al.29 |
| ACTIVATE trial (mitapivat) | Phase 3, adults with PK deficiency, not regularly transfused (<4 transfusions in prior 12 months) and at least one missense PKLR mutation | • 16/40 (40%) receiving mitapivat met the primary endpoint (Hb ≥1.5 g/dL, at least 2 timepoints) compared with 0/40 (0%) receiving placebo • Hemoglobin increase correlated with improvements in markers of hemolysis and hematopoiesis and disease-specific patient reported outcome measures. • Well-tolerated; adverse events (most common: nausea, headache) were similar in the placebo and mitapivat arms. | Al-Samkari H, Galactéros F, Glenthøj A, et al.30 |
| ACTIVATE-T trial (mitapivat) | Phase 3, adults with PK deficiency receiving ≥6 transfusions in prior 12 months and at least 1 missense PKLR mutation | • 10/27 (37%) of adults receiving mitapivat with ≥33% reduction in transfusion burden. • 6/27 (22%) transfusion free after starting mitapivat. • Adverse events included headache, nausea, increase in liver enzymes. | Glenthøj A, van Beers EJ, Al-Samkari H, et al.31 |
| ACTIVATE/ACTIVATE-T open label long-term extension study (mitapivat) | Extension study of ACTIVATE/ACTIVATE-T trials | • ACTIVATE: 39.5% (15/38) randomized to placebo had a Hb response after switching to mitapivat; response by hemoglobin, hemolytic markers, and patient-reported outcomes has been sustained for more than 3 years in the majority. • ACTIVATE-T: 37% (10/27) have met criteria for transfusion response for more than 3 years; 6 patients have remained transfusion free for more than 3 years. • Well-tolerated, no new safety findings. • Decrease in iron overload by liver iron concentration and other iron markers seen in both studies with ongoing improvements over time. • Sustained stability in bone mineral density by DXA scan. | Multiple references32-36 |
DXA, dual energy x-ray absorptiometry; TEAE, treatment-emergent adverse event.
Two phase 1 studies assessed the pharmacodynamics, pharmacokinetics, and safety of mitapivat.26 In the multiple ascending dose healthy volunteer study, the maximum ATP increase was 60% and the maximum decrease in 2,3-BPG was 47%, consistent with activation of PK and the glycolytic pathway. The safety data of both phase 1 studies were reassuring, with no serious treatment-emergent adverse events. The safety and pharmacodynamic data provided support for a mitapivat trial in adults with PK deficiency.
The safety and efficacy of mitapivat in adults with PK deficiency who were not receiving regular transfusions, defined as <4 transfusions in the prior 12 months, were evaluated in the open-label randomized phase 2 DRIVE-PK trial.29 Patients were randomized to 50 mg or 300 mg of mitapivat twice daily for a 24-week period with an optional long-term extension. The primary endpoint of this study was assessment of safety. The therapy was well-tolerated; the most common reported adverse events—headache, insomnia, and nausea—tended to be transient and resolved within 1 week of drug initiation. One patient developed acute hemolysis when mitapivat was stopped, leading to a recommendation for drug taper rather than abrupt discontinuation. In this trial, 50% (26/52) of patients had a hemoglobin increase ≥1 g/dL, with a mean maximum increase of 3.4 g/dL (range: 1.1-5.8 g/dL). The hemoglobin increase occurred quickly (median response in 10 days), with a sustained hemoglobin response seen in the majority with continued dosing and with durable improvements in markers of hemolysis and hematopoiesis. There was a relationship between hemoglobin response and PKLR genotype: all patients with a hemoglobin response had at least 1 missense mutation, whereas the patients with 2 non-missense mutations had poor or no hemoglobin responses (Figure 4).29 None of the 5 patients who were homozygous for the R479H mutation, which is most common in the Amish population, had a hemoglobin response. PK protein levels also correlated with hemoglobin response, consistent with the mechanism of PK activators to bind and stimulate PK.28,29
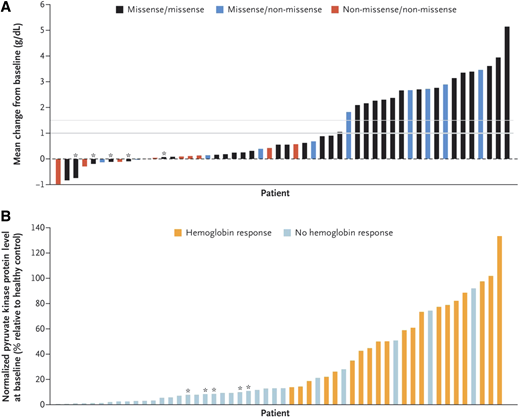
Change in hemoglobin according to PKLR genotype and hemoglobin response according to PK protein level at baseline in the DRIVEPK trial. (A) The mean change from baseline in the hemoglobin level according to the patient's PKLR genotype category. Of the 52 patients who received mitapivat, 20 (38%)—all of whom had at least 1 missense mutation—had a mean hemoglobin increase >1 g/dL (indicated by a thick gray line); 19 patients had a mean hemoglobin increase ≥1.5 g/dL (thin gray line). A mean hemoglobin increase >1 g/dL did not occur in any of the 5 patients who were homozygous for the R479H mutation (as indicated by an asterisk) or in any of the 10 patients who were homozygous for non-missense mutations (as indicated by a red bar). (B) Hemoglobin response according to PK protein level at baseline. Hemoglobin response is defined as >1 g/dL increase in the hemoglobin at >50% of the assessments during the core period. Baseline pyruvate kinase protein level in red cells has been normalized to the hemoglobin level (an approximation of the total red-cell protein level) and to the pyruvate kinase protein level measured in a healthy control (to allow for interassay comparisons). Patients with an increased baseline level of PK protein were more likely to have a hemoglobin response to mitapivat. The data bars in panels A and B are not aligned for each patient, so a 1:1 comparison of the individual data bars in the 2 panels is not possible. From The New England Journal of Medicine, RF Grace et al., Safety and Efficacy of Mitapivat in Pyruvate Kinase Deficiency, 381(1):933-944. Copyright 2019 Massachusetts Medical Society. Reprinted with permission.
Change in hemoglobin according to PKLR genotype and hemoglobin response according to PK protein level at baseline in the DRIVEPK trial. (A) The mean change from baseline in the hemoglobin level according to the patient's PKLR genotype category. Of the 52 patients who received mitapivat, 20 (38%)—all of whom had at least 1 missense mutation—had a mean hemoglobin increase >1 g/dL (indicated by a thick gray line); 19 patients had a mean hemoglobin increase ≥1.5 g/dL (thin gray line). A mean hemoglobin increase >1 g/dL did not occur in any of the 5 patients who were homozygous for the R479H mutation (as indicated by an asterisk) or in any of the 10 patients who were homozygous for non-missense mutations (as indicated by a red bar). (B) Hemoglobin response according to PK protein level at baseline. Hemoglobin response is defined as >1 g/dL increase in the hemoglobin at >50% of the assessments during the core period. Baseline pyruvate kinase protein level in red cells has been normalized to the hemoglobin level (an approximation of the total red-cell protein level) and to the pyruvate kinase protein level measured in a healthy control (to allow for interassay comparisons). Patients with an increased baseline level of PK protein were more likely to have a hemoglobin response to mitapivat. The data bars in panels A and B are not aligned for each patient, so a 1:1 comparison of the individual data bars in the 2 panels is not possible. From The New England Journal of Medicine, RF Grace et al., Safety and Efficacy of Mitapivat in Pyruvate Kinase Deficiency, 381(1):933-944. Copyright 2019 Massachusetts Medical Society. Reprinted with permission.
The safety and efficacy of mitapivat for adults with PK deficiency were demonstrated in two phase 3 clinical trials, leading to its approval by the US Food and Drug Administration (FDA).30,31 The ACTIVATE trial, a phase 3 placebo-controlled trial, randomized 80 adults with PK deficiency who were not receiving regular transfusions to placebo or mitapivat (5 mg to 50 mg twice daily).30 Patient eligibility required at least 1 missense PKLR mutation. The primary endpoint was a hemoglobin increase of ≥1.5 g/dL at 2 or more assessments, a higher threshold for response than the DRIVE-PK study. Mitapivat was well- tolerated, with similar overall adverse event rates in the drug and placebo groups. Of the 40 patients receiving mitapivat, 16 (40%) met the primary endpoint of hemoglobin response, vs 0% in the placebo arm (Figure 5). Additionally, mitapivat decreased hemolysis as measured by change from baseline in indirect bilirubin, lactate dehydrogenase, and haptoglobin and improved hematopoiesis as measured by a reduction in the reticulocyte count. Patient-reported outcomes were measured using 2 disease-specific tools, the PK Deficiency Diary and PK Deficiency Impact Assessment, which were developed to assess quality of life and evaluate the effect of treatment in PK deficiency. Significant and sustained improvements in both measures were seen during the core phase of the trial and have been sustained with continuation of the drug over several years.32
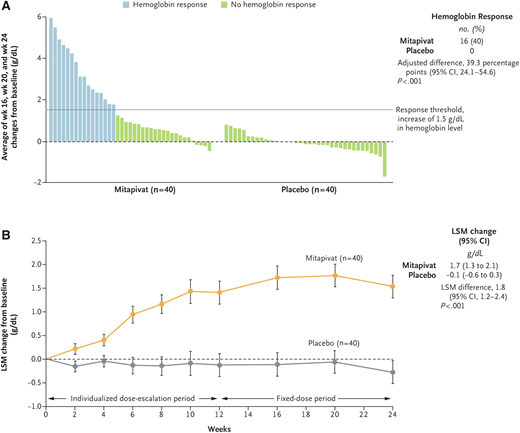
Change in hemoglobin in adults with PK deficiency treated with mitapivat versus placebo on the ACTIVATE T trial. (A) Hemoglobin response, defined as a hemoglobin increase from baseline of ≥1.5 g/dL that was sustained at 2 or more scheduled assessments. Each bar represents an individual patient who was assigned to receive either mitapivat or placebo. (B) The least-squares mean (LSM) change from baseline in the hemoglobin level in the 2 groups during the trial period. The error bars indicate the standard error. From The New England Journal of Medicine, H Al-Samkari et al., Mitapivat versus Placebo for Pyruvate Kinase Deficiency, 386(15):1432-1442.30 Copyright 2022 Massachusetts Medical Society. Reprinted with permission.
Change in hemoglobin in adults with PK deficiency treated with mitapivat versus placebo on the ACTIVATE T trial. (A) Hemoglobin response, defined as a hemoglobin increase from baseline of ≥1.5 g/dL that was sustained at 2 or more scheduled assessments. Each bar represents an individual patient who was assigned to receive either mitapivat or placebo. (B) The least-squares mean (LSM) change from baseline in the hemoglobin level in the 2 groups during the trial period. The error bars indicate the standard error. From The New England Journal of Medicine, H Al-Samkari et al., Mitapivat versus Placebo for Pyruvate Kinase Deficiency, 386(15):1432-1442.30 Copyright 2022 Massachusetts Medical Society. Reprinted with permission.
A phase 3 open-label trial, ACTIVATE-T, evaluated mitapivat in adults with PK deficiency who underwent transfusions regularly (≥6 transfusions in the prior 12 months).31 Mitapivat was well-tolerated, with no major adverse events leading to treatment discontinuation. In this study, 37% (10/27) experienced a reduction in transfusion burden (33% reduction in the number of red cell units transfused during the 24-week core period compared to the historical transfusion burden), and 22% (6/27) achieved a transfusion-free response. Significant and sustained improvements in report of quality of life were also seen for this trial.
Data from the ACTIVATE and ACTIVATE-T open-label extension study have shown sustained benefits with continued administration of mitapivat, including ongoing improvement and stability in hemoglobin, hemolytic markers, markers of hematopoiesis, and patient-reported outcomes.32 -34 Several important long-term benefits have been noted, including a decrease in erythropoiesis and iron overload with improvements in hepcidin, erythropoietin, and liver iron concentration measured by MRI, as compared with placebo in non-transfused adults in the ACTIVATE trial.35 These changes were seen within 6 months of drug initiation and were sustained with continued improvements over time. Long-term follow-up (more than 5 years of monitoring) has also shown general stability of bone mineral density as measured by DXA.36 Although continued follow-up is needed, this stability by DXA in patients with poor bone health prior to trial enrollment suggests that decreasing hemolysis and erythropoiesis with mitapivat may lead to an improvement in bone health.
Long-term data from the phase 2 and phase 3 trials demonstrate favorable safety profiles with more than 5 years of treatment with mitapivat. The phase 3 trials demonstrate that mitapivat has the potential for efficacy in adults with PK deficiency independent of splenectomy or transfusion status. Despite the genotype-hemoglobin response relationship observed in the phase 2 trial, adults with identical PKLR mutations have had different hemoglobin responses to mitapivat. In addition, although patients with 2 non-missense PKLR mutations were excluded from the phase 3 trials, some of these variants have a minor effect on PK protein structure or function, and patients with these variants may experience clinical efficacy with mitapivat. Therefore, a response to treatment cannot be predicted based on genotype. With the recent FDA approval of mitapivat, convincing efficacy in many patients, and a reassuring safety profile, a treatment trial of mitapivat should be considered in all symptomatic adults with PK deficiency except for those individuals who are homozygous for the R479H mutation (Figure 6). Before initiating mitapivat, testing should be performed to determine whether the PK enzyme activity level is low and whether at least 2PKLR mutations of any type are present.
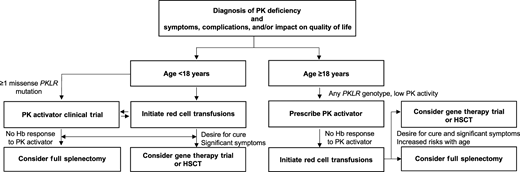
Considerations of treatments for PK deficiency. Supportive and/or disease-targeted treatment should be considered in patients with PK deficiency with symptoms, complications, and/or impact of the disease on quality life. For symptomatic patients ≥18 years, given the reassuring safety profile and favorable clinical efficacy in a substantial subset of adults with PK deficiency, a treatment trial with a PK activator should be initiated. If effective and well-tolerated, the PK activator should be continued with ongoing monitoring. If there is no response to a PK activator, red cell transfusions could be initiated. Hematopoietic stem cell transplant (HSCT) or enrollment in a gene therapy trial could be considered in patients with significant symptoms and/or complications of anemia who are seeking a cure. HSCT outcomes may be better at a younger age. For symptomatic patients <18 years, red cell transfusions should be initiated. Enrollment in a PK activator clinical trial should be considered in those with ≥1 missense PKLR variant and significant symptoms and/or complications of anemia. If there is no response to a PK activator or a patient strongly desires a cure and has significant symptoms, HSCT or enrollment in a gene therapy trial could be considered. Patients should try a PK activator, if available, before proceeding with full splenectomy. Given the complexity of management and treatment decisions in patients with PK deficiency, consideration should be made for a discussion or referral to a hematologist with expertise in PK deficiency. Hemoglobin is indicated by Hb.
Considerations of treatments for PK deficiency. Supportive and/or disease-targeted treatment should be considered in patients with PK deficiency with symptoms, complications, and/or impact of the disease on quality life. For symptomatic patients ≥18 years, given the reassuring safety profile and favorable clinical efficacy in a substantial subset of adults with PK deficiency, a treatment trial with a PK activator should be initiated. If effective and well-tolerated, the PK activator should be continued with ongoing monitoring. If there is no response to a PK activator, red cell transfusions could be initiated. Hematopoietic stem cell transplant (HSCT) or enrollment in a gene therapy trial could be considered in patients with significant symptoms and/or complications of anemia who are seeking a cure. HSCT outcomes may be better at a younger age. For symptomatic patients <18 years, red cell transfusions should be initiated. Enrollment in a PK activator clinical trial should be considered in those with ≥1 missense PKLR variant and significant symptoms and/or complications of anemia. If there is no response to a PK activator or a patient strongly desires a cure and has significant symptoms, HSCT or enrollment in a gene therapy trial could be considered. Patients should try a PK activator, if available, before proceeding with full splenectomy. Given the complexity of management and treatment decisions in patients with PK deficiency, consideration should be made for a discussion or referral to a hematologist with expertise in PK deficiency. Hemoglobin is indicated by Hb.
The approved mitapivat dose ranges from 5 mg to 50 mg twice per day; many patients may require the maximum dose. Hemoglobin response can be evaluated over the first 3-6 months of treatment and is often clear within a few weeks of titration to the maximum dose. Ongoing treatment with the drug is required for a durable effect and should be tapered rather than abruptly discontinued to avoid withdrawal hemolysis. Mitapivat should be avoided in patients who have a sulfa allergy, who have moderate to severe hepatic impairment, and who are also using strong CYP3A inhibitors or inducers. Two mitapivat trials are ongoing for children with PK deficiency. Although there have been no clear implications of the off-target effect of mild aromatase inhibition in adults, this will require careful and ongoing monitoring in children.
CLINICAL CASE (continued)
Treatment options of mitapivat, red cell transfusions, splenectomy, HSCT, and gene therapy clinical trial participation are discussed. The patient initiates mitapivat, and, 1 month after dose titration to 50 mg twice daily, his hemoglobin has increased to 11.5 g/dl. His reticulocyte count is 8%, and his indirect bilirubin is 1.5 mg/dL. He reports a substantial improvement in his energy and has increased his activity level. You start him on oral iron chelation and calcium and vitamin D supplements and refer him to an endocrinologist. He will have regular hematology follow-up appointments to monitor and evaluate this treatment approach.
Conclusions
PK deficiency is a rare chronic hemolytic anemia with variable severity of manifestations, including fatigue, jaundice, iron overload, and decreased patient-reported quality of life. Disease- directed therapy with PK activators is now available for affected adults. Therefore, clinicians must have a high index of suspicion for this disease and send confirmatory diagnostic testing with PK enzyme activity and PKLR genetic sequencing. PK activators have the potential to transform the symptoms and natural history of PK deficiency in many patients by improving hemolytic anemia, hematopoiesis, and quality of life and decreasing the risk of long-term complications. A trial of this medication in symptomatic adults with PK deficiency is warranted; however, mitapivat is not effective for all individuals with PK deficiency. In a subset of patients, often with significant symptoms and complications, there may not be a clear benefit from PK activators. Gene therapy is a promising and potentially curative treatment option currently under development; although this approach may address an unmet need in this subset of patients, the long-term efficacy and safety are under evaluation. Disease-targeted treatment through PK activation and gene therapy are innovative approaches for the management of PK deficiency that may significantly improve clinical manifestations and quality of life of affected patients.
Acknowledgments
The visual abstract and Figure 3 were created using Biorender.
Conflict-of-interest disclosure
Rachael Grace: Research funding to institution (Agios, Sobi, Novartis); consultancy (Agios, Sanofi).
Off-label drug use
Rachael Grace: No discussion of off-label drug use. Nothing to disclose.
