

Abstract
Hemoglobin S (HbS) polymerization, red blood cell (RBC) sickling, chronic anemia, and vaso-occlusion are core to sickle cell disease (SCD) pathophysiology. Pyruvate kinase (PK) activators are a novel class of drugs that target RBC metabolism by reducing the buildup of the glycolytic intermediate 2,3-diphosphoglycerate (2,3-DPG) and increasing production of adenosine triphosphate (ATP). Lower 2,3-DPG level is associated with an increase in oxygen affinity and reduction in HbS polymerization, while increased RBC ATP may improve RBC membrane integrity and survival. There are currently 3 PK activators in clinical development for SCD: mitapivat (AG-348), etavopivat (FT-4202), and the second-generation molecule AG-946. Preclinical and clinical data from these 3 molecules demonstrate the ability of PK activators to lower 2,3-DPG levels and increase ATP levels in animal models and patients with SCD, as well as influence a number of potential pathways in SCD, including hemoglobin oxygen affinity, RBC sickling, RBC deformability, RBC hydration, inflammation, oxidative stress, hypercoagulability, and adhesion. Furthermore, early-phase clinical trials of mitapivat and etavopivat have demonstrated the safety and tolerability of PK activators in patients with SCD, and phase 2/3 trials for both drugs are ongoing. Additional considerations for this novel therapeutic approach include the balance between increasing hemoglobin oxygen affinity and tissue oxygen delivery, the cost and accessibility of these drugs, and the potential of multimodal therapy with existing and novel therapies targeting different disease mechanisms in SCD.
Learning Objectives
Learn the therapeutic mechanisms of action of pyruvate kinase activators in sickle cell disease
Evaluate the existing evidence supporting the use of pyruvate kinase activators in the treatment of sickle cell disease
CLINICAL CASE
A 24-year-old woman with sickle cell disease (SCD; hemoglobin SS), complicated by chronic anemia with a baseline hemoglobin of 6 to 7 g/dL and significantly elevated hemolysis markers, alloimmunization, iron overload, dilated cardiomyopathy with preserved ejection fraction, cholelithiasis requiring cholecystectomy, and occasional acute pain episodes presents to an outpatient clinic for follow-up. She reports chronic fatigue and occasional dyspnea on exertion when walking up hills or flights of stairs. She has been on hydroxyurea for 2 years, with a maximum tolerated dose of 1500 mg daily due to cytopenias. She reports compliance with both hydroxyurea and deferasirox and would like to know if she has any other therapeutic options.
Chronic anemia is a hallmark complication of SCD. Anemia pathophysiology in SCD begins with hemoglobin S (HbS) polymerization and sickling of red blood cells (RBCs), leading to intravascular and extravascular hemolysis, reducing RBC life span and the oxygen delivery capacity of blood, and resulting in tissue hypoxia and chronic ischemic organ damage.1 The focus of therapeutic development to address anemia has targeted strategies to limit HbS polymerization in deoxygenated sickle RBCs, including increasing fetal hemoglobin (HbF) levels, altering hemoglobin oxygen affinity, and RBC hydration. Remarkable clinical improvements have resulted from these efforts, particularly evident in hydroxyurea's ability to increase HbF, reducing microvascular occlusion, tissue ischemia, and pain.
However, chronic anemia remains a major contributor to morbidity and mortality in SCD, and better treatments for anemia have the potential to further improve outcomes in SCD. Low hemoglobin concentration is associated with neurocognitive impairment in SCD, even in the absence of structural abnormalities on magnetic resonance imaging.2 In a recent meta-analysis, a modeled increase in hemoglobin concentration of ≥1 g/dL was associated with a reduction in the risk of cerebrovascular disease, albuminuria, elevated estimated pulmonary artery systolic pressure, and mortality.3 In addition, prominent symptoms of anemia, such as fatigue, are common in individuals living with SCD and greatly affect their quality of life and physical functioning.4,5 However, there are no prospective data demonstrating that raising hemoglobin levels can actually improve cognitive function or clinical outcomes in SCD. At the same time, blood viscosity is abnormally increased in SCD due to decreased sickle RBC deformability, increased adhesion, and abnormal vasoreactivity, such that excessive increases in hemoglobin concentration may lead to viscosity-related complications.1 For example, rapid and dramatic correction of anemia with simple RBC transfusions has anecdotally led to increased intracranial pressure, seizure, and stroke in patients with SCD.6-8 A few epidemiologic studies have also found a correlation between increased hemoglobin levels and increased vaso-occlusive crisis (VOC) frequency,9,10 and a phase 3 study of senicapoc (ICA-17043) was stopped early due to lack of efficacy in terms of reduction of VOCs despite an observed increase in hemoglobin and decrease in hemolysis.11 This highlights a fundamental knowledge gap in the field surrounding the pathogenesis of chronic microvascular tissue ischemia in SCD: which process contributes more to tissue ischemia, the reduction in oxygen content and delivery resulting from anemia or vaso-occlusion in the microcirculation, which may be worsened by increased blood viscosity? Further research is needed to understand how therapies for anemia may influence or balance these competing mechanisms of hypoxia.
While there are limited existing treatment options for chronic anemia in SCD, the therapeutic arena for SCD is rapidly expanding. Blood transfusions are the mainstay therapy for severe anemia but carry risks of iron overload, alloimmunization, hemolytic transfusion reactions, and hyperhemolysis in SCD. Hydroxyurea, the primary drug therapy for SCD, has only a modest effect on hemoglobin level in adults, and patients who do not tolerate or remain anemic despite hydroxyurea need additional therapy. Recombinant human erythropoietin (EPO) is the standard of care for anemia of chronic kidney disease, but there are limited and conflicting data on its efficacy in SCD.12-15 In 2019, voxelotor, a hemoglobin oxygen affinity modifier, was approved for SCD after a phase 3 study demonstrated a ∼1-g/dL increase in hemoglobin in 51% of patients with SCD.16 More recently, early-phase studies of a novel drug class in clinical development, the pyruvate kinase activators, have shown potential for raising hemoglobin level and reducing sickling and hemolysis in individuals with SCD.
RBC energetics in SCD
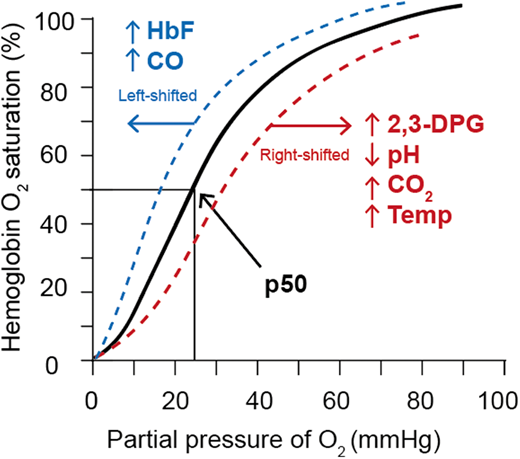
Erythrocytes metabolize glucose through the Embden-Myerhof pathway to produce adenosine triphosphate (ATP), the sole source of energy for the cell. Although seemingly inefficient, the pathway also produces reduced nicotinamide adenine dinucleotide to reduce methemoglobin, glucose-6-phosphate to drive the pentose phosphate shunt to make nicotinamide adenine dinucleotide phosphate to reduce glutathione and protect against oxidation of hemoglobin, cytoskeletal protein, and membrane lipids, as well as the Rapoport-Luebering shunt to generate 2,3-diphosphoglycerate (2,3-DPG; Figure 1).17 2,3-DPG, H+, CO2, CO, temperature, and HbF all affect the oxygen affinity of hemoglobin (p50, ie, the partial pressure of oxygen at which 50% of the hemoglobin is saturated with oxygen; Figure 2).17 An increase in 2,3-DPG shifts the oxygen dissociation curve to the right and increases p50, while a decrease in 2,3-DPG decreases p50. Thus, increased levels of 2,3-DPG in SCD increase p50 and shift the oxygen dissociation curve to the right, favoring HbS deoxygenation and polymerization. In the last step of glycolysis, phosphoenolpyruvate (PEP) is catalyzed to pyruvate by pyruvate kinase (PK) and accounts for 50% of the RBC ATP production. Depletion of ATP in SCD leads to insufficient ATP for proper functioning of ATP-dependent ion pumps such as the Na+/K+ pump, causing defective ion transport, and Ca++ influx through Piezo1 activates the Gardos channel, resulting in K+ outflux and RBC dehydration, increased intracellular HbS concentration, and promotion of HbS deoxygenation and polymerization.18
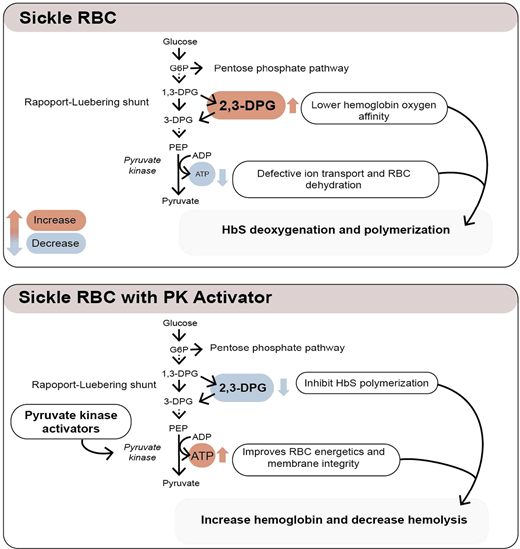
PK activation as a potential therapeutic approach in SCD
PK deficiency is the most common enzyme deficiency causing nonspherocytic hemolytic anemia and iron overload. PK-deficient RBCs have increased 2,3-DPG and decreased ATP, with the oxygen dissociation curve shifted to the right.19 Similarly, increased levels of 2,3-DPG and decreased RBC ATP are seen in sickle RBCs,20,21 causing an increase in the p50 of hemoglobin, HbS polymerization, and sickling, as well as RBC dehydration. Additionally, a recent study shows less PK stability and activity in sickle than control RBCs.22 Furthermore, coinheritance of PK deficiency and sickle cell trait may induce sickling, causing an SCD phenotype.23,24 There are 290 pyruvate kinase liver and red blood cell (PKLR) mutations, with 276 being pathogenic.17 Genetic variants of PKLR have shown to be associated with acute pain in patients with SCD25 and, together with the expected metabolic effects, provide a potential rationale for the use of PK activators as a therapeutic strategy in SCD.
The tetrameric R form of red cell PK (PKR) has low affinity for PEP and can be increased by binding of fructose-1,6-bisphosphate (FBP), the major allosteric activator of PKR. FBP binding acts to increase PEP binding affinity, promoting tetramerization and stabilizing the PKR enzyme in the tetrameric state.26 Recently, mitapivat and etavopivat (Figure 3) were developed as oral allosteric activators to bind a pocket at the dimer-dimer interface distinct from the FBP-binding domain. This results in an increase in PKR activity in wild-type and mutant enzymes. Mitapivat (AG-348) has since been approved by the US Food and Drug Administration (FDA) and shown in PK-deficient patients to significantly increase the hemoglobin level, decrease hemolysis, and improve patient-reported outcomes.27 Thus, targeting activation of PKR in sickle RBCs to lower 2,3-DPG and increase ATP levels seems like a reasonable strategy to increase hemoglobin and decrease sickling and hemolysis.
CLINICAL CASE (continued)
Various treatment options are reviewed with the patient, including EPO and 3 recently approved drug therapies for SCD: voxelotor (a modifier of hemoglobin oxygen affinity), L-glutamine (an antioxidant), and crizanlizumab (a P-selectin inhibitor). Due to her heavy alloimmunization and severe iron overload, chronic transfusion therapy is not recommended. Given her severe anemia and high hemolytic rate, a clinical trial of a pyruvate kinase activator is also considered.
Preclinical and clinical studies of PK activators in SCD
Mitapivat
Mitapivat activates both wild-type and mutant forms of the PK enzyme and has been evaluated for the treatment of PK deficiency, SCD, and thalassemia, as well as approved by the FDA for PK deficiency in 2022. An ex vivo study of mitapivat treatment on RBCs from patients with SCD found an increase in ATP and decreases in 2,3-DPG, p50, and point of sickling (a measure of hypoxia-induced sickling using oxygen gradient ektacytometry).22 Findings from a preclinical study of mitapivat in the SCD (HbSS) Townes mouse model were mixed; in contrast to human data, the HbSS mice had higher PKR protein and ATP and lower 2,3-DPG compared to the control (HbAA) mice.28 Mitapivat did not significantly impact 2,3-DPG or hemoglobin levels but did further increase ATP levels and decrease spleen size, leukocytosis, RBC reactive oxygen species levels, and RBC mitochondrial retention in HbSS mice, suggesting potential beneficial mechanisms apart from inhibition of HbS polymerization by reducing 2,3-DPG.
Completed phase 1 and phase 2 single-center multiple ascending dose studies (NCT04000165, EudraCT 2019-003438-18; Table 1) demonstrated promising clinical effects of mitapivat in patients with SCD.29,30 Pharmacokinetic and pharmacodynamic effects of mitapivat were demonstrated to be similar to data from healthy volunteers, with a dose-dependent increase in serum drug and ATP levels and a dose-dependent decrease in 2,3-DPG levels. In the phase 1 study,29 participants with SCD treated with mitapivat had a significant increase in mean hemoglobin of 1.2 g/dL, and 56.3% of participants achieved a ≥1-g/dL increase in hemoglobin from baseline. Mean reductions in hemolytic markers were observed as well. Furthermore, there was a trend toward decreased p50 and increased time to sickling, t50; while not significant, these findings support the mechanism of action of decreasing 2,3-DPG to increase hemoglobin oxygen affinity and delay sickling. The phase 2 study (ESTIMATE) showed similar results, with a mean increase in hemoglobin level of 1.3 g/dL and 1.1 g/dL during the 8-week dose finding and the 1-year fixed-dose extension period, respectively,30,31 as well as a significant increase in ATP and reduction in 2,3-DPG, hemolytic markers, p50, and point of sickling during the extension period, presented as a conference abstract.31
Summary of ongoing and completed clinical trials of pyruvate kinase activators in sickle cell disease
| Trial | Subjects | Study design | Status | |
|---|---|---|---|---|
| Mitapivat (AG-348) | NCT0400016529 | N = 17, age ≥18, HbSS | Phase 1, open-label, multiple ascending dose study | Completed |
| ESTIMATE (EudraCT 2019-003438-18)30,31 | N = 9, age ≥16, HbSS, HbS/β0- or HbS/β+-thalassemia | Phase 2, open-label, multiple ascending dose phase, followed by fixed-dose extension study | Ongoing | |
| NCT04610866 | N = 15, age 18-70, HbSS | Phase 1/2, open-label extension study | Ongoing | |
| RISE UP (NCT05031780) | N = 267, age ≥16, any SCD genotype | Phase 2/3, randomized, placebo-controlled, double-blind, followed by open-label extension study | Ongoing | |
| Etavopivat (FT-4202) | NCT0381569534-36 | N = 130, healthy volunteers and patients with SCD (any genotype) age 12-65 | Phase 1, randomized, placebo-controlled, double-blind, single ascending and multiple ascending dose study | Completed |
| NCT04987489 | N = 60, age 12-65, patients with thalassemia and any SCD genotype | Phase 2, open-label study | Ongoing | |
| NCT05725902 | N = 12, age 12-21, HbSS or HbS/β0 | Phase 2, open-label study | Ongoing | |
| HIBISCUS (NCT04624659) | N = 344, age 12-65, any SCD genotype | Phase 2/3, randomized, placebo-controlled, double-blind, followed by open-label extension study | Ongoing | |
| HIBISCUS-KIDS (PACTR202209604592389) | N = 50, age 12 to <18, any SCD genotype | Phase 1/2, open-label, single-arm, followed by extension study | Ongoing | |
| AG-946 | NCT04536792 | N = 64, age 18-70, healthy volunteers and patients with SCD (any genotype) | Phase 1, open-label, single ascending and multiple ascending dose study | Ongoing |
| Trial | Subjects | Study design | Status | |
|---|---|---|---|---|
| Mitapivat (AG-348) | NCT0400016529 | N = 17, age ≥18, HbSS | Phase 1, open-label, multiple ascending dose study | Completed |
| ESTIMATE (EudraCT 2019-003438-18)30,31 | N = 9, age ≥16, HbSS, HbS/β0- or HbS/β+-thalassemia | Phase 2, open-label, multiple ascending dose phase, followed by fixed-dose extension study | Ongoing | |
| NCT04610866 | N = 15, age 18-70, HbSS | Phase 1/2, open-label extension study | Ongoing | |
| RISE UP (NCT05031780) | N = 267, age ≥16, any SCD genotype | Phase 2/3, randomized, placebo-controlled, double-blind, followed by open-label extension study | Ongoing | |
| Etavopivat (FT-4202) | NCT0381569534-36 | N = 130, healthy volunteers and patients with SCD (any genotype) age 12-65 | Phase 1, randomized, placebo-controlled, double-blind, single ascending and multiple ascending dose study | Completed |
| NCT04987489 | N = 60, age 12-65, patients with thalassemia and any SCD genotype | Phase 2, open-label study | Ongoing | |
| NCT05725902 | N = 12, age 12-21, HbSS or HbS/β0 | Phase 2, open-label study | Ongoing | |
| HIBISCUS (NCT04624659) | N = 344, age 12-65, any SCD genotype | Phase 2/3, randomized, placebo-controlled, double-blind, followed by open-label extension study | Ongoing | |
| HIBISCUS-KIDS (PACTR202209604592389) | N = 50, age 12 to <18, any SCD genotype | Phase 1/2, open-label, single-arm, followed by extension study | Ongoing | |
| AG-946 | NCT04536792 | N = 64, age 18-70, healthy volunteers and patients with SCD (any genotype) | Phase 1, open-label, single ascending and multiple ascending dose study | Ongoing |
Most treatment-related adverse events were nonserious in both studies. The phase 1 study reported 2 VOCs as related to drug, 2 VOCs not related to drug, and a serious adverse event (SAE) of pulmonary embolism not related to drug.29 The first VOC that was possibly related to drug occurred during the drug taper period, leading to a protocol amendment to extend the drug taper length. The second possibly drug-related VOC also occurred during drug taper, although in the setting of self- reported VOC triggers. The ESTIMATE study reported a massive pulmonary embolism due to COVID-19 resulting in death and a grade 4 SAE of urinary tract infection with hypotension that were both unrelated to drug.30,31 During the extension period, 4 VOCs occurred, with 3 in the setting of documented noncompliance the week before. The mean annualized VOC rate and SCD-related hospital admission days were decreased compared to baseline, although these differences were not significant.30,31 Overall, the 2 studies concluded that mitapivat was safe and tolerable in participants with SCD, and a phase 2/3 multicenter study, RISE UP (Table 1), is in progress, with primary end points of hemoglobin response, treatment-emergent adverse events (phase 2), and annualized rate of VOC (phase 3).
Etavopivat
Etavopivat (FT-4202) is another small-molecule PK activator in clinical development for SCD and thalassemia. Preclinical studies of etavopivat in the Berkeley sickle cell anemia (BERK SCA) mouse model showed significantly decreased 2,3-DPG and increased ATP levels, which were associated with decreases in p50, point of sickling, number of irreversibly sickled cells on blood smear, and increased RBC deformability.32 In addition, treatment with etavopivat led to a mean hemoglobin increase of 1.7 g/dL, a significant reduction in hemolytic markers, and a 28.5% increase in RBC half-life compared to untreated BERK SCA mice, demonstrating clear mechanistic benefits of decreasing 2,3-DPG and increasing ATP in sickle RBCs. Ex vivo studies similarly showed a significant decrease in p50 and point of sickling with etavopivat treatment of RBCs from patients with HbSS and HbSC disease.33
While the full results of a completed phase 1/2 study of etavopivat in SCD (NCT03815695; Table 1) have not yet been published, results to date have been presented as conference abstracts.34-36 In the 12-week open-label extension phase, the mean maximal hemoglobin increase from baseline was 1.5 g/dL, with 73.3% achieving a hemoglobin increase of >1 g/dL.34 Increases in hemoglobin and decreases in hemolytic markers were significant at all time points between 2 and 12 weeks of treatment.
In line with the etavopivat preclinical and mitapivat clinical data, 2,3-DPG decreased and ATP increased from baseline throughout the 12 weeks of treatment, followed by a return to baseline after 4 weeks of washout.35 A significant decrease in mean p50, point of sickling, and dense RBCs and an increase in RBC deformability were observed at 12 weeks of treatment.34 Additionally, data from the 2-week and 12-week treatment phases of the study demonstrated an increased mean corpuscular volume, decreased mean corpuscular hemoglobin concentration, increased antioxidant capacity, and decreased markers of inflammation, hypercoagulability, adhesion, and serum erythropoietin level, suggesting potential pleiotropic salutary effects of PK activation in SCD.34,35
Most safety events were grades 1 to 2, most commonly sickle cell pain events and headache.34,36 On study, there were 2 SAEs of VOC unrelated to study drug and an SAE of left femoral vein deep vein thrombosis possibly related to study drug. Compared to historical data, the annualized rate of VOC requiring hospitalization during the 12-week treatment period was lower, although this difference was not significant. A phase 2/3 randomized, placebo-controlled study (HIBISCUS; Table 1) in adults and adolescents with SCD and a phase 1/2 open-label study (HIBISCUS- KIDS) in children with SCD are ongoing, with primary end points of hemoglobin response and annualized VOC rate (HIBISCUS) and pharmacokinetic assessments plus safety and tolerability (HIBISCUS-KIDS), respectively.
AG-946
AG-946 is a second-generation PK activator in clinical development for the treatment of SCD and low-risk myelodysplastic syndrome. AG-946 differs pharmacokinetically and pharmacodynamically from first-generation mitapivat in that AG-946 has greater potency, a longer half-life (∼80-110 hours vs 3-6 hours), and more prolonged effects on 2,3-DPG and ATP levels (observed at least 14 days after last dose in healthy controls),37 which provides a potential self-tapering effect. Preclinical data presented in a conference abstract demonstrated AG-946's ability to normalize levels of glycolytic intermediates, decrease 2,3-DPG levels, and increase hemoglobin levels in a Townes HbSS mouse model, although it did not significantly affect ATP levels or degree of RBC sickling.38 Ex vivo treatment of RBCs from patients with SCD with AG-946 also decreased p50 and point of sickling, similar to first-generation PK activators.39 In an ongoing phase 1 clinical trial, enrollment in single and multiple ascending dose cohorts for healthy volunteers has been completed and was deemed to be safe; this study is now enrolling multiple sequential dosing cohorts in participants with SCD (NCT04536792).
CLINICAL CASE (continued)
The patient elects to participate in a phase 2/3 clinical trial of a pyruvate kinase activator. Her hemoglobin level, hemolytic markers, and rate of VOC will be monitored closely.
Concerns about increasing hemoglobin oxygen affinity
Recent concerns were raised about the importance of oxygen delivery by RBCs in anemia and drugs that affect the oxygen affinity of hemoglobin.40 Individuals with a normal hemoglobin level have baseline 20% hemoglobin oxygen unloading, while in anemia, 30% is unloaded due to increased levels of 2,3-DPG. This compensatory effect can be observed in PK deficiency, which markedly shifts the oxygen dissociation curve to the right through increased 2,3-DPG, allowing for more oxygen unloading; exercise tolerance on a bicycle ergometer was greater in a PK-deficient individual than an individual with hexokinase deficiency, with a similar hemoglobin but lower 2,3-DPG level. The article reminds hematologists of this basic physiologic principle when considering PK activators or voxelotor, drugs that alter oxygen affinity but increase hemoglobin by a modest 1 to 2 g/dL. It is important to keep in mind that compared with other types of anemias, the lowering of 2,3-DPG through PK activation may increase the delay time in HbS polymerization and prevent sickling, providing a uniquely beneficial effect in SCD. Further concerns have been raised about cerebral oxygen delivery with drugs that alter oxygen affinity in SCD,41 with conflicting evidence in the literature; 1 recent conference abstract showed that voxelotor treatment in children with SCD reduces cerebral metabolic stress by improving cerebral oxygen delivery,42 while another abstract showed no changes in cerebral blood flow and oxygen metabolic rate (CMRO2), despite increased cerebral oxygen delivery due to improvement of hemoglobin levels.43 These data reflect the complex physiology underlying cerebral hemodynamics and metabolism in SCD, which requires further investigation.
Additional considerations
While the preclinical and clinical data to date for PK activators are promising, additional placebo-controlled studies and demonstration of effects on clinical outcomes such as VOC frequency are needed in the SCD population. There has also been some concern over withdrawal of therapeutic effect with abrupt cessation of therapy. In patients with PK deficiency, treatment cessation at higher doses of mitapivat led to episodes of withdrawal hemolysis and anemia, prompting the addition of a drug taper, with no further episodes of withdrawal hemolysis on study.44 While there was no clear evidence of withdrawal hemolysis associated with the VOCs occurring during the clinical trials in SCD, a sudden stop in drug dosing due to noncompliance or side effects could theoretically translate into an abrupt increase in 2,3-DPG levels and a consequent shift of the oxygen dissociation curve to the right, promoting an acute increase in deoxy-HbS polymerization, sickling, and vaso-occlusion. On the other hand, increased RBC ATP may improve RBC survival, which may provide a longer-term protective effect. The results of the ongoing phase II/III studies of mitapivat and etavopivat should shed additional light on this question.
Another consideration is the cost of novel therapies and global access to these therapies. Insurance approval has proven a barrier to access for recently approved drug therapies for SCD in the United States, and the out-of-pocket cost has been prohibitive for the use of these new drugs in low- and middle-income countries (LMICs). As drug development advances in the SCD arena, more effort needs to be focused on equitable distribution of drug access to LMICs, where most of the SCD disease burden is concentrated.
Finally, with multiple drug therapies now available for SCD and numerous novel therapies coming down the pipeline, further research into how to compare, combine, or sequence drug therapies is needed to establish data-driven guidelines for the treatment of complications in SCD. With existing and potential drugs targeting HbF induction, hemoglobin oxygen affinity, RBC metabolism, hemolysis, adhesion, oxidative stress, and complement activation, we are approaching a reality in which multimodal drug therapy will hopefully usher in an era of precision medicine in SCD,45 providing targeted treatment of different disease phenotypes.
Acknowledgments
This work was supported in part by the National Institutes of Health Grant 5KL2TR001856. Figure 1 and visual abstract illustrations by Dr. Lydia Perkins.
Conflict-of-interest disclosure
Julia Z. Xu is the US national principal investigator for the phase 1 clinical trial of AG-946 in patients with sickle cell disease. Julia Z. Xu receives research funding and serves on an advisory committee for GlaxoKlineSmith.
Gregory M. Vercellotti receives research funding from CLS Behring, Omeros, and Novartis and serves on the DSMB of Novo Nordisk/Forma, Alexion, Hillhurst, and Sangamo.
Off-label drug use
Julia Z. Xu: There is nothing to disclose.
Gregory M. Vercellotti: There is nothing to disclose.
