

Abstract
Thrombocytopenia in ill children is common; accurately diagnosing the underlying etiology is challenging and essential for appropriate management. Triggers for accelerated consumption of platelets are numerous; common downstream mechanisms of clearance include platelet trapping in microvascular thrombi, phagocytosis, and platelet activation. Thrombocytopenia with microangiopathic hemolytic anemia (MAHA) is frequently due to disseminated intravascular coagulation. Thrombotic microangiopathy (TMA) is a subgroup of MAHA. Specific TMA syndromes include thrombotic thrombocytopenic purpura, complement-mediated TMA (CM-TMA), and Shiga toxin–mediated hemolytic uremic syndrome. Isolated thrombocytopenia is characteristic of immune thrombocytopenia; however, concomitant cytopenias are frequent in critically ill patients, making the diagnosis difficult. Immune thrombocytopenia with large vessel thrombosis is a feature of heparin-induced thrombocytopenia and antiphospholipid antibody syndrome. In addition, thrombocytopenia is common with macrophage activation, which is characteristic of hemophagocytic lymphohistiocytosis. While thrombocytopenia in ill patients can be driven by hypoproliferative processes such as myelosuppression and/or bone marrow failure, this review will focus on consumptive thrombocytopenia due to immune and nonimmune causes.
Learning Objectives
To understand the mechanisms that contribute to consumptive thrombocytopenia in ill pediatric patients
To know how to distinguish between different etiologies for consumptive thrombocytopenia
Approach to consumptive thrombocytopenia
Thrombocytopenia in ill children is often multifactorial, and the pathophysiology may be overlapping. Identification of specific etiologies for consumptive thrombocytopenias can be challenging. Consumptive thrombocytopenia may be immune mediated, either due antibodies (immune thrombocytopenia [ITP]) or secondary to T-cell activation and/or phagocytosis (eg, hemophagocytic lymphohistiocytosis [HLH]). Microangiopathic hemolytic anemia (MAHA) is a common nonimmune mechanism of thrombocytopenia that can arise from overlapping syndromes of disseminated intravascular coagulation (DIC) and thrombotic microangiopathies (TMAs). Specific characteristics of these disorders and can help distinguish between diagnoses:
Schistocytes—MAHA, TMA
Renal dysfunction/hypertension/proteinuria— complement mediated TMA (CM-TMA), hemolytic uremic syndrome (HUS)
Severe coagulopathy—DIC
Young age—inherited genetic mutations ADAM metallopeptidase with thrombospondin type 1 motif 13 [ADAMTS13]—thrombotic thrombocytopenic purpura [TTP]; complement regulatory factors—TMA; effector T cell function or perforin trafficking—HLH)
Significant hyperferritinemia—HLH
Bloody diarrhea—Shiga toxin–mediated HUS (ST-HUS)
Severe and isolated thrombocytopenia—ITP
Thrombosis— heparin-induced thrombocytopenia (HIT; with heparin exposure), antiphospholipid antibody syndrome (APLS)
When evaluating an ill child with thrombocytopenia, applying diagnostic tools and systematically assessing diagnostic criteria can aid the identification of specific syndromes. Table 1 highlights shared and distinct clinical and laboratory features of these syndromes.
Comparison of syndromes with microangiopathic hemolytic anemia (MAHA) with thrombocytopenia
| Features | DIC | Thrombotic microangiopathies (TMAs) | HLH | ||
|---|---|---|---|---|---|
| TTP | ST-HUS | CM-TMA | |||
| Etiology | Tissue factor–mediated thrombin activation | ADAMTS13 deficiency | Shiga toxin–induced endothelial injury | Complement activation; deficiency of complement inhibitors | CD8 T-cell activation |
| Pathology | Systemic microvascular thrombosis | Systemic microvascular thrombosis | Renal microvascular thrombosis | Renal microvascular thrombosis | INF-γ-mediated macrophage activation |
| Acquired causes | Infection, malignancy, trauma, vascular tumors, circuits | Autoantibodies to ADAMTS13; autoimmune disease | Shiga toxin from E. coli | Autoantibodies to complement proteins, HSCT, drug, pregnancy | Malignancy, autoimmune disease, rheumatologic disease |
| Genetic variants | Neonatal purpura fulminans: PROC | ADAMTS13 | None | Complement regulatory proteins: CFH, CD46, CFI, C3, CFB, THBD | Perforin trafficking and effector T-cell function: PRF1, UNC13D, STX11, STXBP2, Rab27A, SH2D1A, BIRC4, ITK |
| Renal involvement Other organ involvement | Variable Multiorgan dysfunction | Infrequent Brain | Frequent; AKI, proteinuria, HTN Brain | Frequent; AKI, proteinuria, HTN Lung, gastrointestinal system, brain, serositis | Variable Multiorgan dysfunction, hepatosplenomegaly, |
| Laboratory screening | ADAMTS13 nl, low; sC5b-9 nl; D dimer very high; ferritin high | ADAMTS13, very low; sC5b-9 nl D-dimer nl, high Ferritin nl, high | ADAMTS13 nl; sC5b-9 high; D-dimer nl, high; ferritin nl, high | ADAMTS13 nl; sC5b-9 high; D-dimer nl; Ferritin nl, high | ADAMTS13 nl; sC5b-9 nl; D dimer, high; ferritin, very high; sCD25, very high; CXCL9, very high |
| Diagnosis | ISTH DIC score ≥5 | ADAMTS13 < 10% | E. coli 0157:H7 in stool | TMA diagnostic criteria39 | HLH diagnostic criteria38 |
| Treatment | Treat primary cause | Plasma exchange; immunosuppression | Supportive; anti-complement considered with neurologic symptoms | Anti-complement therapy | Immunosuppression; Anti-T cell therapy; HSCT |
| Features | DIC | Thrombotic microangiopathies (TMAs) | HLH | ||
|---|---|---|---|---|---|
| TTP | ST-HUS | CM-TMA | |||
| Etiology | Tissue factor–mediated thrombin activation | ADAMTS13 deficiency | Shiga toxin–induced endothelial injury | Complement activation; deficiency of complement inhibitors | CD8 T-cell activation |
| Pathology | Systemic microvascular thrombosis | Systemic microvascular thrombosis | Renal microvascular thrombosis | Renal microvascular thrombosis | INF-γ-mediated macrophage activation |
| Acquired causes | Infection, malignancy, trauma, vascular tumors, circuits | Autoantibodies to ADAMTS13; autoimmune disease | Shiga toxin from E. coli | Autoantibodies to complement proteins, HSCT, drug, pregnancy | Malignancy, autoimmune disease, rheumatologic disease |
| Genetic variants | Neonatal purpura fulminans: PROC | ADAMTS13 | None | Complement regulatory proteins: CFH, CD46, CFI, C3, CFB, THBD | Perforin trafficking and effector T-cell function: PRF1, UNC13D, STX11, STXBP2, Rab27A, SH2D1A, BIRC4, ITK |
| Renal involvement Other organ involvement | Variable Multiorgan dysfunction | Infrequent Brain | Frequent; AKI, proteinuria, HTN Brain | Frequent; AKI, proteinuria, HTN Lung, gastrointestinal system, brain, serositis | Variable Multiorgan dysfunction, hepatosplenomegaly, |
| Laboratory screening | ADAMTS13 nl, low; sC5b-9 nl; D dimer very high; ferritin high | ADAMTS13, very low; sC5b-9 nl D-dimer nl, high Ferritin nl, high | ADAMTS13 nl; sC5b-9 high; D-dimer nl, high; ferritin nl, high | ADAMTS13 nl; sC5b-9 high; D-dimer nl; Ferritin nl, high | ADAMTS13 nl; sC5b-9 nl; D dimer, high; ferritin, very high; sCD25, very high; CXCL9, very high |
| Diagnosis | ISTH DIC score ≥5 | ADAMTS13 < 10% | E. coli 0157:H7 in stool | TMA diagnostic criteria39 | HLH diagnostic criteria38 |
| Treatment | Treat primary cause | Plasma exchange; immunosuppression | Supportive; anti-complement considered with neurologic symptoms | Anti-complement therapy | Immunosuppression; Anti-T cell therapy; HSCT |
ADAMTS13, ADAM metallopeptidase with thrombospondin type 1 motif 13; CM-HUS, complement-mediated hemolytic uremic syndrome; DIC, disseminated intravascular coagulation; E. coli, Escherichia coli; HLH, hemophagocytic lymphohistiocytosis; HSCT, hematopoietic stem cell transplant; HTN, hypertension; ISTH, International Society on Thrombosis and Haemostasis; nl, normal; ST-HUS, Shiga toxin–mediated hemolytic uremic syndrome; TMA, thrombotic microangiopathy; TTP, thrombotic thrombocytopenic purpura.
CLINICAL CASE #1
A 15-year-old male presented a left ilio-femoral deep vein thrombus (DVT) (Figure 1A). Therapeutic anticoagulation with unfractionated heparin (UFH) was initiated, and he underwent catheter-directed thrombolysis. Seven days after initiation of UFH, he developed increased leg swelling and shortness of breath; imaging showed progression of the lower extremity DVT and a pulmonary embolus (PE). Progressive thrombocytopenia was also noted with a platelet count of 358 000 per mm3 on admission that dropped to a nadir of 54 000 per mm3. A 4T (Thrombocytopenia, Timing, Thrombosis, and oTher etiologies) score showed a high probability of heparin-induced thrombocytopenia (HIT) (Figure 1B). UFH was discontinued, and he was transitioned to the direct thrombin inhibitor (DTI) bivalirudin. A platelet factor 4 (PF4) immunoassay was strongly positive, and confirmatory serotonin release assay (SRA) was also positive. He continues long-term anticoagulation with rivaroxaban.
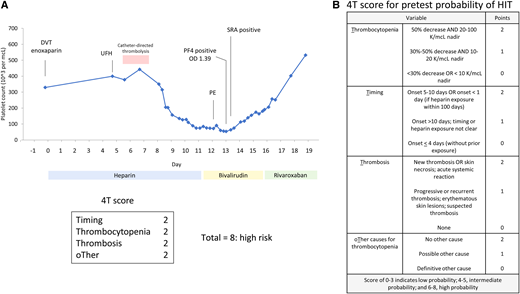
Clinical case 1: heparin induced thrombocytopenia (HIT). (A) Time course for platelet count (blue) with heparin exposure demonstrates characteristic pattern of HIT. The platelet factor 4 (PF4) immunoassay was strongly positive (OD = 1.39; cutoff for positivity OD ≥0.4), and the serotonin release assay (SRA) was positive. Platelet count recovered after discontinuation of heparin and transition to a direct thrombin inhibitor, bivalirudin, and then rivaroxaban. (B) 4T score for pretest probability of HIT. The patient scored a high probability of HIT with a score of 8. DVT, deep vein thrombos; OD, optical density; PE, pulmonary embolism; UFH, unfractionated heparin.
Clinical case 1: heparin induced thrombocytopenia (HIT). (A) Time course for platelet count (blue) with heparin exposure demonstrates characteristic pattern of HIT. The platelet factor 4 (PF4) immunoassay was strongly positive (OD = 1.39; cutoff for positivity OD ≥0.4), and the serotonin release assay (SRA) was positive. Platelet count recovered after discontinuation of heparin and transition to a direct thrombin inhibitor, bivalirudin, and then rivaroxaban. (B) 4T score for pretest probability of HIT. The patient scored a high probability of HIT with a score of 8. DVT, deep vein thrombos; OD, optical density; PE, pulmonary embolism; UFH, unfractionated heparin.
Heparin-induced thrombocytopenia
This patient presented with worsening thrombosis and new onset thrombocytopenia during heparin exposure. The development of thrombocytopenia with venous or arterial thrombosis should raise the suspicion of HIT. Heparin-induced thrombocytopenia is a prothrombotic, drug-associated immune thrombocytopenia with significant morbidity and risk of mortality.1 It is characterized by the development of platelet activating IgG antibodies that target platelet factor 4 (PF4)/heparin complexes. The antibodies typically develop within 5 to 10 days of heparin exposure, which correlate with onset of thrombocytopenia (Figure 2). Paradoxically, HIT is associated with venous and arterial thrombosis; bleeding is uncommon. Heparin use and thrombocytopenia are common in critically ill patients; however, the confirmed diagnosis of HIT is rare. HIT is more common with UFH but can be seen with low molecular weight heparin (LMWH). The rate of HIT in adults2 varies from 0.1% to 7%; in children,3 the rate is lower at 0.046%, although the reported incidence from smaller studies varies.4,5 Clinical evaluation and appropriate laboratory testing are important to prevent overdiagnosis and unnecessary exposure to alternative anticoagulants.6
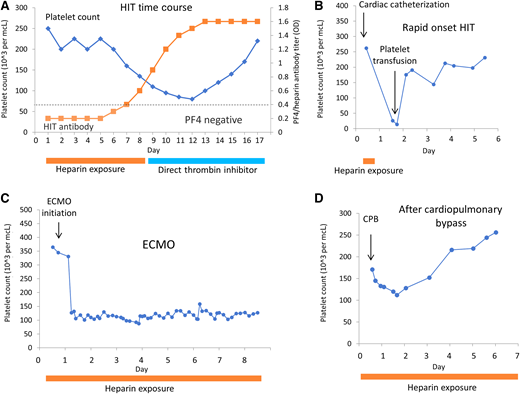
Patterns of thrombocytopenia for HIT and conditions confused for HIT. (A) Time course for drop in platelets (blue) in response to increase in HIT antibody titer (orange squares). Recovery of platelet count with discontinuation of heparin and transition to direct thrombin inhibitor. (B) Rapid onset HIT. Patient was previously treated with heparin and developed rapid onset of thrombocytopenia with heparin exposure. (C) Thrombocytopenia with extracorporeal membrane oxygenation (ECMO)—not HIT. Rapid and sustained thrombocytopenia is seen with initiation of ECMO due to platelet consumption within the circuit. (D) Thrombocytopenia after cardiopulmonary bypass (CPB)—not HIT. Platelet drop within 1-3 days after CPB, followed by recovery.
Patterns of thrombocytopenia for HIT and conditions confused for HIT. (A) Time course for drop in platelets (blue) in response to increase in HIT antibody titer (orange squares). Recovery of platelet count with discontinuation of heparin and transition to direct thrombin inhibitor. (B) Rapid onset HIT. Patient was previously treated with heparin and developed rapid onset of thrombocytopenia with heparin exposure. (C) Thrombocytopenia with extracorporeal membrane oxygenation (ECMO)—not HIT. Rapid and sustained thrombocytopenia is seen with initiation of ECMO due to platelet consumption within the circuit. (D) Thrombocytopenia after cardiopulmonary bypass (CPB)—not HIT. Platelet drop within 1-3 days after CPB, followed by recovery.
The diagnosis of HIT is made clinically and supported with laboratory testing. The 4T is a validated predictive scoring system that determines the pretest probability of HIT. This score incorporates 4 Ts to characterize the likelihood of HIT (Figure 1B).7 A low 4T score has a high negative predictive value in both adults and children and can be used to rule out HIT.3,4,8 Recognition of characteristic patterns of thrombocytopenia with heparin exposure is key to the diagnosis; the onset of thrombocytopenia accompanies antibody formation (5-10 days), unless there was previous exposure to heparin (rapid- onset HIT) (Figure 2A, B). Examples of other conditions with heparin exposure that can be confused as HIT, such as cardiopulmonary bypass and extracorporeal membrane oxygenation, are illustrated in Figure 2C and D.
Laboratory testing for HIT antibodies is needed to support the diagnosis and include immunoassays and functional assays. ELISA-based immunoassays for PF4/heparin complexes are widely available with rapid turnaround time for results. Only a small fraction of patients that test positive by immunoassays will have a clinical diagnosis of HIT. A positive immunoassay should be followed up with functional testing, such as performing the serotonin release assay (SRA). In adults, a negative PF4 ELISA assay (OD value <0.4) has a high negative predictive value; thus, a negative result is useful to rule out HIT. The utility of the PF4 assay has been evaluated in children, and a low titer similarly has a high negative predictive value for HIT.3,4
However, the PF4 immunoassays has a high false positive rate and should only be sent with an intermediate or high 4T score.2 If the immunoassay returns positive, then the SRA should be performed to confirm the diagnosis. If the 4T score is high, then heparin should be discontinued and patients should be transitioned to a nonheparin anticoagulant, such as intravenous bivalirudin or argatroban, subcutaneous fondaparinux, or a direct oral anticoagulant (DOAC).2 DOACs have been used exclusively to treat adult patients with HIT. In children, the DOACs rivaroxaban and dabigatran have recently been approved by the US Food and Drug Administration for treatment of thrombosis; however, the experience with their use in HIT is limited. For patients with an intermediate-risk 4T score, the degree of PF4 positivity can be useful to assess the likelihood of HIT; higher OD values are associated with increased risk of HIT.9,10 Functional SRA testing is used to confirm or rule out HIT in these indeterminate cases; however, the turnaround time for results may be several days, as this testing is typically available only through reference laboratories. Recently, a nonradioactive platelet-activation assay, the PF4-dependent P-selectin expression assay, has shown comparable accuracy to SRA for confirming HIT. This test is technically simple to perform and may facilitate the timely diagnosis of HIT.11
Immune thrombocytopenia due to secondary causes in ill children
Most children with ITP are clinically well and do not need hospitalization. However, some will present with other significant symptoms associated with underlying diagnoses or triggers. Heparin-induced thrombocytopenia is an uncommon drug- associated immune thrombocytopenia with significant morbidity and requires recognition, appropriate testing, and specific management. The finding of large vessel venous or arterial thrombosis with thrombocytopenia after heparin exposure, as illustrated in case 1, is a hallmark of HIT. The development of thrombosis with ITP can also be seen with APLS. Other clinical conditions that may be associated ITP should prompt the provider to consider associated diagnoses. Multilineage cytopenias can be caused by both hypoproliferative and consumptive processes. Immune thrombocytopenia with autoimmune hemolytic anemia and/or immune neutropenia was previously described as Evans syndrome. Many individuals with this syndrome of multilineage immune cytopenias have acquired autoimmune diseases or underlying inherited immunoregulatory disorders.12
CLINICAL CASE #2
A 14-year-old female presented with fatigue and headaches and was diagnosed with mononucleosis. Laboratory evaluation showed severe and isolated thrombocytopenia. Urinalysis was positive for hemoglobin, and she was treated presumptively for ITP with intravenous immunoglobulin (IVIG) (Figure 3A). The platelet count did not improve, and she developed worsening anemia with increased markers of hemolysis; schistocytes were identified on peripheral blood smear. The diagnosis of TTP was considered, and a prediction score for TTP (PLASMIC score, Figure 3B) showed a high probability of TTP. She was started on steroids and plasmapheresis; subsequently, her thrombocytopenia improved. Her ADAMTS13 activity was undetectable with high titer inhibitor, consistent with acquired TTP due to an autoantibody.
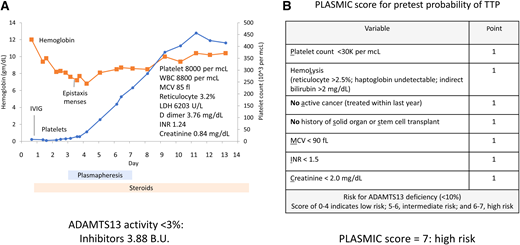
Clinical case 3: thrombotic thrombocytopenic purpura (TTP). (A) Patient presented with severe and selective thrombocytopenia (blue). Platelets did not improve after intravenous immunoglobulin (IVIG) treatment for presumptive immune thrombocytopenia (ITP). Patient developed a progressive decrease in hemoglobin (orange), and markers of hemolysis consistent with a microangiopathic anemia. Platelet count normalized with plasmapheresis and steroids. INR, international normalized ratio; LDH, lactate dehydrogenase; MCV, mean corpuscular hemoglobin; WBC, white blood cells. (B) PLASMIC score was high, compatible with TTP.
Clinical case 3: thrombotic thrombocytopenic purpura (TTP). (A) Patient presented with severe and selective thrombocytopenia (blue). Platelets did not improve after intravenous immunoglobulin (IVIG) treatment for presumptive immune thrombocytopenia (ITP). Patient developed a progressive decrease in hemoglobin (orange), and markers of hemolysis consistent with a microangiopathic anemia. Platelet count normalized with plasmapheresis and steroids. INR, international normalized ratio; LDH, lactate dehydrogenase; MCV, mean corpuscular hemoglobin; WBC, white blood cells. (B) PLASMIC score was high, compatible with TTP.
Thrombotic thrombocytopenic purpura
This patient presented with isolated thrombocytopenia suggestive of ITP. She subsequently developed MAHA without renal dysfunction, and TTP was suspected. TTP is caused by a deficiency of the von Willebrand factor–cleaving protease, ADAMTS13. ADAMTS13 cleaves ultra large von Willebrand factor (ULVWF) into smaller multimers; in the absence of ADAMTS13, these ULVWF can bind to platelets and accumulate in the microvasculature, resulting in accelerated platelet clearance. In contrast to other TMAs, TTP is not typically associated with acute kidney injury. The incidence in adults is 2.88 cases per million, while in children the incidence is 0.09 cases per million.13 In children, TTP may be acquired due to inhibitory antibodies or may be hereditary secondary to biallelic mutations in the ADAMTS13 gene. Hereditary TTP commonly presents with neonatal hyperbilirubinemia with hemolytic anemia and thrombocytopenia; the median age of diagnosis is 5.5 years.14 It can be misdiagnosed as ITP or neonatal alloimmune thrombocytopenia. Acquired TTP is more common in females and is associated with autoimmune diseases such as systemic lupus erythematosus. Neurologic symptoms are frequent in patients with hereditary and acquired TTP and can range from headaches and lethargy (as seen in this patient) to stroke.14
The PLASMIC score (Figure 3B) was high in this patient, identifying this patient at high risk for TTP and severely low ADAMTS13 activity (<10%).15 This clinical prediction tool can assist treatment decision-making, given the variable availability and turnaround times for ADAMTS13 results. The PLASMIC score uses creatine for renal function assessment; however, creatine varies with age in the pediatric population. Recently, the PLASMICkid score was proposed with similar criteria as the PLASMIC score but uses estimated glomerular filtration rate to account for the changing normal creatinine levels in children.16 This score has been evaluated in a single institution and requires validation in a larger cohort.
Hereditary TTP is treated with fresh frozen plasma infusions, whereas acquired TTP is managed with both plasmapheresis and immunosuppression with steroids and rituximab.17 A humanized monoclonal antibody to von Willebrand factor, caplacizumab, has been developed and prevents interaction of the platelets with ULVWF. It has been shown to be effective in improving thrombocytopenia in acquired-TTP treatment.18,19 Experience with caplacizumab in the pediatric population is limited; however, successful outcomes have been published in case reports and case series.20
Other thrombotic microangiopathies
While the main driver of TTP is an ADAMTS13 deficiency, other TMAs are triggered by endothelial injury and characterized by complement dysregulation. The most common TMAs encountered in the pediatric population include CM-TMA and ST-HUS or other infections. Other etiologies include drug-induced TMA, metabolic TMA (associated with cobalamin deficiency), vasculitis-associated TMA (associated with systemic lupus erythematosus, APLS, and anti-neutrophil cytoplasmic antibodies) and hypertension-associated TMA.21,22
These disorders are characterized by MAHA with microvascular thrombosis typically involving the renal vasculature with acute kidney injury; however, organ involvement can be widespread. ST-HUS is associated with bloody diarrhea and is caused by Shiga toxin–induced endothelial injury. ST-HUS and other infection-associated HUSs are typically managed with supportive care. CM-TMA describes a group of inherited and acquired TMAs caused by genetic mutations in complement genes or autoantibodies to complement regulatory proteins. Complement activation leads to endothelial damage with subsequent end-organ damage. In patients who present with features typical of HUS without bloody diarrhea, hereditary HUS should be considered. This condition is due to mutations in genes that encode complement proteins (Table 1). Another increasingly recognized type of CM-TMA is hematopoietic stem cell transplant (HSCT) associated TMA (TA-TMA). TA-TMA is a severe complication of HSCT triggered by endothelial injury. Genetic variants for complement regulatory factors as well as autoantibodies are described in this population.23,24 Biopsies show characteristic histologic changes, but biopsy is not required for CM-TMA diagnosis. Evaluation of complement activity (C3, C4, CH50, sC5b-9) can aid in the diagnosis, with elevations of sC5b-9 supportive of the diagnosis (Table 1). If there is there is high suspicion of CM-TMA, then anticomplement therapy (eg, eculizumab, an anti-C5 antibody) should be initiated.25-27
CLINICAL CASE #3
A 16-year old male with cerebral palsy was hospitalized after tendon release surgery. Postoperatively, he developed fever and pneumonia. Additional findings included leukocytosis with anemia, thrombocytopenia, and coagulopathy (Figure 4A). Schistocytes were noted on peripheral blood smear. The DIC score was 6, consistent with diagnosis of DIC (Figure 4B). He was also encephalopathic; renal function was normal. A PLASMIC score showed an intermediate probability of TTP. Because of the unexplained encephalopathy, he underwent plasmapheresis, which was discontinued when the ADAMTS13 level returned as normal. He improved with antibiotics for pneumonia and supportive care.
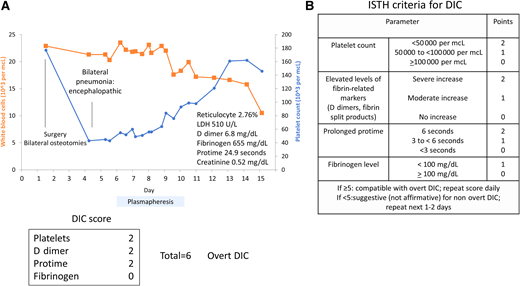
Clinical case 3: disseminated intravascular coagulation (DIC). (A) Patient with pneumonia develops a rapid drop in platelet count (blue) and increased white count (orange) with microangiopathic hemolytic anemia and high D dimer. Thrombotic thrombocytopenic purpura (TTP) was considered given neurologic symptoms, and plasmapheresis was initiated. The patient recovered with antibiotic treatment for pneumonia and sepsis. (B) International Society on Thrombosis and Haemostasis (ISTH) disseminated intravascular coagulation (DIC) score high, compatible with overt DIC.
Clinical case 3: disseminated intravascular coagulation (DIC). (A) Patient with pneumonia develops a rapid drop in platelet count (blue) and increased white count (orange) with microangiopathic hemolytic anemia and high D dimer. Thrombotic thrombocytopenic purpura (TTP) was considered given neurologic symptoms, and plasmapheresis was initiated. The patient recovered with antibiotic treatment for pneumonia and sepsis. (B) International Society on Thrombosis and Haemostasis (ISTH) disseminated intravascular coagulation (DIC) score high, compatible with overt DIC.
Disseminated intravascular coagulation
This patient presented with an acute illness associated with precipitous drop in platelet count. The findings of schistocytes on smear with markers of hemolysis are consistent with MAHA. The most common etiology for MAHA is DIC, although other thrombotic microangiopathies, such as TTP, should be considered. Significant elevations of the INR and/or depression of fibrinogen, as seen in this patient, are more typical of DIC rather than TTP and other TMAs (Table 1). These diagnoses may overlap and may be difficult to distinguish; because of unexplained mental status changes, he was empirically treated for TTP until the ADAMTS13 activity returned to normal.
DIC is an acquired condition that causes extensive activation of the coagulation system. DIC is not a specific disease, but a physiologic process in response to an illness or injury that has become pathologic. It is precipitated by numerous conditions, such as sepsis, trauma, malignancy, mechanical circuits, and vascular tumors; the common trigger is tissue factor–mediated thrombin generation.28 Accompanying this process is an inability to dampen thrombin activity due to a decrease in the natural anticoagulants antithrombin, protein C, and protein S. The fibrinolytic pathway can also be inhibited with an increase in plasminogen activator inhibitor 1. Dysregulated thrombin activation results in consumption of prothrombotic factors with fibrin deposition in small vessels and platelet trapping.29 Depletion of platelets and coagulation factors predispose a person to bleeding, and microthrombi contribute to progressive organ dysfunction.30 While DIC is typically an acquired condition, biallelic mutations in PROC, the gene encoding protein C, manifests as DIC with purpura fulminans at birth.31
Several scoring systems for diagnosis and assessing severity of DIC were harmonized by the International Society on Thrombosis and Haemostasis.32 Increased severity is characterized by worsening thrombocytopenia, prolongation of prothrombin time, reduction in fibrinogen levels, and elevation of fibrin-split products and D dimer. A score of ≥5 is compatible with overt DIC, and <5 is suggestive of nonovert DIC.32,33 In children, age-dependent coagulation factor changes may affect the performance of DIC scores; however, several studies have demonstrated their utility to predict morbidity and mortality in the pediatric population.34-36 Treatment of DIC is focused on treating the underlying condition. Replacement of coagulation factors is usually not indicated in the absence of active bleeding.37
Hemophagocytic lymphohistiocytosis
Summary
Mechanisms underlying consumptive thrombocytopenia include antibody-mediated destruction, phagocytosis, and microangiopathy. Appropriate and prompt diagnosis is important, as disease-specific therapies may be lifesaving. Heparin- induced thrombocytopenia is critical to recognize and treat, but appropriate diagnostic tools and laboratory interpretation are essential to prevent overdiagnosis. Microangiopathic hemolytic anemia syndromes have considerable overlap and may be difficult to distinguish. Management of DIC is directed at treating underlying etiologies. However, TTP requires specific management with plasmapheresis, and CM-TMA should be managed with anticomplement therapy. Hemophagocytic lymphohistiocytosis may have overlapping features with these syndromes and is treated with immunosuppression. Distinguishing these diagnoses can be challenging; a systematic approach to identification of specific etiologies can aid diagnosis and is critical to implement appropriate treatment.
Conflict-of-interest disclosure
Rohith Jesudas: no competing financial interests to declare.
Clifford M. Takemoto: research funding from GBT, Forma Therapeutics, and Daiichi Sankyo; Novartis Data Safety Monitoring Committee.
Off-label drug use
Rohith Jesudas: enoxaparin, bivalirudin, eculizamab, caplacizumab.
Clifford M. Takemoto: enoxaparin, bivalirudin, eculizamab, caplacizumab.
