

Abstract
Leukocytosis is a common finding in pediatric patients, and the differential diagnosis can be broad, including benign reactive leukocytosis and malignant myeloproliferative disorders. Transient abnormal myelopoiesis is a myeloproliferative disorder that occurs in young infants with constitutional trisomy 21 and somatic GATA1 mutations. Most patients are observed, but outcomes span the spectrum from spontaneous resolution to life-threatening complications. Juvenile myelomonocytic leukemia is a highly aggressive myeloproliferative disorder associated with altered RAS-pathway signaling that occurs in infants and young children. Treatment typically involves hematopoietic stem cell transplantation, but certain patients can be observed. Early recognition of these and other myeloproliferative disorders is important and requires a clinician to be aware of these diagnoses and have a clear understanding of their presentations. This paper discusses the presentation and evaluation of leukocytosis when myeloproliferative disorders are part of the differential and reviews different concepts regarding treatment strategies.
Learning Objectives
To describe the differential diagnosis of leukocytosis in a young child
To recognize the signs and symptoms consistent with an underlying myeloproliferative disorder
To discuss treatment options for TAM and JMML
Introduction
Leukocytosis is a common condition for which pediatric hematology/oncology providers are consulted. Leukocytosis is defined as an elevated white blood cell (WBC) count for age (Table 1). The term leukemoid reaction describes a WBC count >50 × 103/µL with neutrophils and immature myeloid forms typically occurring in the setting of an (often pyogenic) infection, while hyperleukocytosis typically refers to a WBC count >100 × 103/µL.1 Leukocytosis may be caused by an elevation in any WBC subtype, and the differential diagnosis for leukocytosis can often be narrowed based on which cell type is elevated (Figure 1).1,2 Leukocytosis can be a primary disease process (such as in malignant or myeloproliferative disorders) or occur secondary to an identifiable underlying etiology (eg, infection).
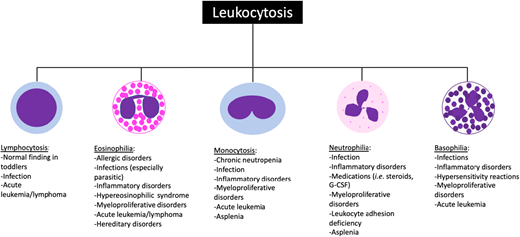
Common causes of leukocytosis in pediatric patients. The differential diagnosis can often be narrowed down based on which white blood cell subtype is elevated. G-CSF, granulocyte colony stimulating factor.
Common causes of leukocytosis in pediatric patients. The differential diagnosis can often be narrowed down based on which white blood cell subtype is elevated. G-CSF, granulocyte colony stimulating factor.
Normal white blood cell ranges by age
| Age | Total white blood cells ( × 103/µL) Mean (± 2SD) |
|---|---|
| 1-3 days | 18.9 (9.4-34) |
| 2 weeks | 11.4 (5-20) |
| 1 month | 10.8 (4-19.5) |
| 6 months-2 years | 10.6 (6-17) |
| 2-6 years | 8.5 (5-15.5) |
| 6-12 years | 8.1 (4.5-13.5) |
| 12-18 years | 7.8 (4.5-13.5) |
| Age | Total white blood cells ( × 103/µL) Mean (± 2SD) |
|---|---|
| 1-3 days | 18.9 (9.4-34) |
| 2 weeks | 11.4 (5-20) |
| 1 month | 10.8 (4-19.5) |
| 6 months-2 years | 10.6 (6-17) |
| 2-6 years | 8.5 (5-15.5) |
| 6-12 years | 8.1 (4.5-13.5) |
| 12-18 years | 7.8 (4.5-13.5) |
Adapted from The Harriet Lane Handbook, 23rd Edition.
The workup for pediatric patients with leukocytosis depends on their presentation. All patients require a thorough history and physical examination. A complete blood count (CBC) with differential and peripheral blood smear are important to evaluate the WBC differential and morphology as well as other hematologic parameters (including hemoglobin and platelet count). Additional workup may be required depending on the results of these evaluations and clinical suspicion for a primary versus a secondary disease process.
CASE 1
You are consulted by the neonatal intensive care unit (NICU) regarding a 5-day-old full-term infant with trisomy 21 (T21) found to have leukocytosis on a CBC sent during an infectious workup. The patient's clinical history is notable for a prenatal diagnosis of T21; the postnatal course has been notable for respiratory distress transiently requiring positive pressure and a low-grade fever prompting the recent infectious workup. Physical examination is notable for stigmata of T21 and hepatomegaly. Review of the patient's laboratory results reveals leukocytosis with WBC 60.8 × 103/µL and 40% blasts, with normal hemoglobin and platelet counts. The patient has been started on broad-spectrum antibiotics and pediatric hematology/oncology providers are consulted for evaluation of leukocytosis.
Approaching this patient
This patient is a neonate presenting with fever and leukocytosis. Infection—particularly serious bacterial infection—is an important consideration in any infant presenting with fever, and infants may have a left-shift with immature forms on peripheral smear in this setting. However, in this infant, the increased peripheral blast count is suggestive of a more serious underlying etiology. Key to this case is the patient's history of T21 (Down syndrome; DS): since these patients have an increased risk of developing myeloproliferative disorders and acute leukemia at young ages, these disorders should be high on the differential diagnosis of leukocytosis in an infant with T21.
Abnormal myelopoiesis in trisomy 21
Patients with T21 are at significantly increased risk for leukemia: while the overall risk of acute leukemia in patients with T21 is 18-fold higher than in the general population, the risk of acute myeloid leukemia in T21 patients aged <5 years is almost 400 times greater.3,4 Abnormal myelopoiesis in patients with T21 is driven by mutations in the hematopoietic transcription factor GATA1 and comprises 2 conditions: transient abnormal myelopoiesis (TAM) and myeloid leukemia of Down syndrome (ML-DS), both of which are classified by the World Health Organization (WHO) as “myeloid proliferations associated with Down syndrome.”5 Although these conditions share a genetic driver, the clinical features and management differ, and both must be considered in a patient with T21 and leukocytosis.
Transient abnormal myelopoiesis
Transient abnormal myelopoiesis (previously transient myeloproliferative disorder) is a clonal hematopoietic disorder unique to patients with T21 (Table 2). TAM is characterized by an increase in circulating blast cells harboring somatic mutations in GATA1.6,7 Without appropriate GATA1 signaling, normal hematopoietic development is disrupted, leading to the phenotype seen in TAM.8 TAM has been estimated to occur in 10%-30% of infants with T21.9 Infants with GATA1 mutations who have blast percentages <10% and lack the clinical features of classic TAM have been designated as having silent TAM.8
Key features of TAM and JMML
| TAM | JMML | |
|---|---|---|
| Clinical features | Diagnosis of trisomy 21 | Male to female ratio 2:1 |
| Age ≤3 months | Most common in early childhood | |
| Jaundice and/or HSM are common | Splenomegaly in almost all patients | |
| Rash and effusions are less frequent | Lymphadenopathy and fever are common | |
| Laboratory findings | Leukocytosis | Leukocytosis |
| Circulating blasts (higher in blood than BM) | Monocytosis (≥1 × 109/L) | |
| Blast count <20% in blood and BM | ||
| Anemia is rare | Thrombocytopenia is common | |
| Genetic features | GATA1 mutation | Germline mutations/LOH: CBL, NF1 |
| Or | ||
| Somatic mutations: PTPN11, KRAS, NRAS, RRAS |
| TAM | JMML | |
|---|---|---|
| Clinical features | Diagnosis of trisomy 21 | Male to female ratio 2:1 |
| Age ≤3 months | Most common in early childhood | |
| Jaundice and/or HSM are common | Splenomegaly in almost all patients | |
| Rash and effusions are less frequent | Lymphadenopathy and fever are common | |
| Laboratory findings | Leukocytosis | Leukocytosis |
| Circulating blasts (higher in blood than BM) | Monocytosis (≥1 × 109/L) | |
| Blast count <20% in blood and BM | ||
| Anemia is rare | Thrombocytopenia is common | |
| Genetic features | GATA1 mutation | Germline mutations/LOH: CBL, NF1 |
| Or | ||
| Somatic mutations: PTPN11, KRAS, NRAS, RRAS |
BM , bone marrow; HSM , hepatosplenomegaly; JMML , juvenile myelomonocytic leukemia; LOH , loss of heterozygosity; TAM , transient abnormal myelopoiesis.
Patients with TAM typically present within the first week of life, but may be diagnosed up to 3 months of age.10 While ∼25% of patients with TAM are clinically asymptomatic at presentation, patients more commonly present with a constellation of symptoms caused by organ infiltration of malignant cells, including hepatomegaly, pleural/pericardial effusions, and skin rash.10,11 Laboratory findings characteristically include leukocytosis and an increased peripheral blast count exceeding that of the marrow; hemoglobin is typically normal, and platelet count is usually normal or slightly reduced.10,11 Patients may also demonstrate abnormal liver function and/or coagulation testing due to hepatic infiltration. Peripheral smear shows pleiomorphic circulating blast cells with a unique megakaryocytic immunophenotype (Figure 2A).12 Bone marrow evaluation is not required to diagnose TAM but is often done in the setting of peripheral blasts to rule out leukemia.
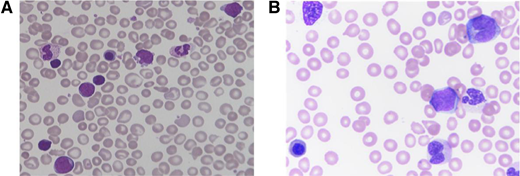
Peripheral blood findings in TAM and JMML. Peripheral blood smears of (A) TAM showing pleiomorphic blasts and additional immature myeloid cells, and (B) JMML showing leukoerythroblastosis with circulating myelomonocytic cells. Both smears show dysmorphic and nucleated red blood cells.
Peripheral blood findings in TAM and JMML. Peripheral blood smears of (A) TAM showing pleiomorphic blasts and additional immature myeloid cells, and (B) JMML showing leukoerythroblastosis with circulating myelomonocytic cells. Both smears show dysmorphic and nucleated red blood cells.
Most patients with TAM experience spontaneous resolution of symptoms without intervention. In a prospective study by the Children's Oncology Group, 79% of patients experienced resolution of peripheral blasts in a median of 36 days and of all TAM symptoms in a median of 49 days without treatment.10 A smaller percentage of patients with TAM require intervention, typically with low-dose cytarabine. However, there is no consensus on which patients should receive treatment: some have reported improved survival by treating patients with poor prognostic features including leukocytosis or liver dysfunction, while others reserve treatment for patients with life-threatening symptoms.10,13,14 Following symptom resolution, patients with TAM require close monitoring, as 16%-23% will develop ML-DS.10,11,13 Treatment with cytarabine has not been shown to prevent progression from TAM to ML-DS, although data suggest that systemic steroids may be beneficial.15,16
Myeloid leukemia of Down syndrome
ML-DS occurs in patients with a history of TAM or silent TAM when the aberrant GATA1 clone acquires additional oncogenic mutations.17,18 This leads to a megakaryoblastic leukemia that is distinct from the one occurring in non-DS patients.12,19 ML-DS onset is typically at 1-2 years of age (almost always before 4 years), and patients typically present with progressive pancytopenia.20 Patients with ML-DS are treated with cytotoxic chemotherapies at reduced doses; this is because the leukemic blasts of ML-DS patients show increased chemotherapy sensitivity and because patients with T21 suffer increased toxic morbidity from standard- dose chemotherapies.21,22 Treatment regimens for ML-DS typically include anthracyclines and cytarabine, though there is disagreement about dosing intensity: a recent Children's Oncology Group study suggests that high-dose cytarabine is necessary, while Japanese studies have shown similar outcomes using lower-dose therapy.22,23 The prognosis for patients with ML-DS is highly favorable with survival rates >85%, although patients with relapsed or refractory disease have poor outcomes.21-24
Infantile myeloproliferative disease
It has recently been recognized that a rare subset of neonates without Down syndrome can present with a transient myeloproliferative disorder, termed infantile myeloproliferative disease (IMD).25 Though the number of reported IMD cases is small, recurrent mutations have been identified, including somatic T21 and GATA1. While IMD can often be managed with a “watch and wait” approach, it is important to differentiate these cases from more aggressive leukemias, as has been described in recent consensus guidelines.25
Differential considerations
Patients with T21 presenting with leukocytosis should always be evaluated for infection, as these patients often have immune dysregulation.26 Patients with T21 are also at higher risk for acute lymphoblastic leukemia (DS-ALL) than the general pediatric population, although this is quite rare in young infants.8 DS-ALL demonstrates unique biologic features compared with non-DS-ALL, and patients with DS-ALL experience worse outcomes—driven both by higher treatment-related mortality and higher rates of relapse—than non-DS-ALL patients.27,28 Additional conditions that may cause nonspecific findings similar to those seen in TAM, such as hepatomegaly, should also be ruled out.
CASE 1 (outcome)
The patient was given a presumptive diagnosis of TAM. Given the patient's good clinical status and lack of life-threatening symptoms, the decision was made for close monitoring without intervention. The patient was discharged from the NICU once the infectious workup was negative and was followed closely by pediatric hematologists/oncologists. Serial CBCs revealed resolution of peripheral blasts and normalization of WBC count within 4 weeks. The patient is now 2 years old and continues to be closely monitored due to the ongoing risk of progression to ML-DS, though all recent CBCs have been normal.
CASE 2
An 8-week-old boy with a history of a 1-week NICU admission for respiratory distress is admitted to the pediatric inpatient unit for evaluation and management of a left neck abscess. The patient was initially noted to have dysmorphic features during his NICU course, including low-set ears, down-slanting palpebral fissures, cryptorchidism, and an abnormal hearing test; genetic testing for Noonan syndrome is pending. A CBC reveals WBC 41.3 × 103/µL, hemoglobin 10 g/dL, and platelet count 65 × 103/µL; the differential blood count is notable for neutrophilia (absolute neutrophil count 19.4 × 103/µL) and monocytosis (absolute monocyte count 11.2 × 103/µL). A review of prior CBCs reveals persistent leukocytosis with monocytosis and thrombocytopenia since birth. Pediatric hematologist/oncologist are consulted for evaluation for leukocytosis.
Approaching this patient
When thinking about potential causes for this child's leukocytosis, it is reasonable to attribute a left-shifted, increased WBC count to an infection. However, this infant has had leukocytosis since birth, which is concerning for an underlying disorder. In addition, while thrombocytopenia could be due to infection, it could also be due to an underlying marrow disorder. A critical component of this child's history and exam is the finding of dysmorphic features concerning for Noonan syndrome and the presence of abnormal blood counts since birth. The suspicion of a myeloproliferative disorder should be high in a child with an underlying since many genetic conditions are associated with an increased risk of developing myeloid malignancies.
Juvenile myelomonocytic leukemia
Juvenile myelomonocytic leukemia (JMML) is a clonal hematopoietic disorder characterized by excessive proliferation of granulocytic and monocytic lineage cells.29 It was previously classified by the WHO as a myeloproliferative/myelodysplastic overlap syndrome but is now recognized as a distinct myeloproliferative condition of childhood in both the recently-updated WHO diagnostic criteria and the newly described International Consensus Classification system.5,30 Overactive RAS-pathway signaling drives JMML, and the majority of patients (∼90%) are found to have alterations in a RAS-pathway gene, including PTPN11, NRAS, KRAS, CBL, and NF1.30
JMML predominantly affects infants and young children, with a median age of 1.8 years at diagnosis.31 The incidence of JMML is estimated between 0.69 and 1.3 per million children per year, approximately 2%-3% of pediatric hematologic malignancies,32,33 and is twice as common in males.31 Clinical features of JMML are caused by organ infiltration by malignant monocytic cells. Presenting symptoms commonly include pallor, fever, infection, bleeding, cough, and malaise, while physical findings include hepatosplenomegaly (in most patients), lymphadenopathy, skin rash, café au lait spots, and xanthomas.31 Peripheral smear demonstrates profound monocytosis, often with dysplasia, circulating myeloid precursors, and erythroblasts (Figure 2B).33 Bone marrow findings often include hypercellularity with a predominance of myeloid lineage cells at all stages of maturation, dysplasia which can be seen across all hematopoietic lineages, and occasionally reticulin fibrosis.33-35
The diagnostic criteria for JMML are detailed by the WHO and the International Consensus Classification system and require both clinical and genetic findings.5,30 Clinical and lab features include splenomegaly, monocytosis, and peripheral and bone marrow blast counts below 20% (Table 2). Somatic alterations consistent with JMML include mutations in PTPN11, KRAS, NRAS, or RRAS. Germline heterozygous mutations in NF1 (or a neurofibromatosis diagnosis) or CBL can lead to loss of heterozygosity in the bone marrow in JMML, most commonly through a mechanism of uniparental disomy. It is critical to distinguish between germline and somatic mutations, particularly in the setting of PTPN11, NRAS, or KRAS mutations, which cause Noonan syndrome when present in the germline but can be consistent with JMML when present as somatic alterations. Skin or marrow fibroblasts are ideal tissue sources for germline testing; buccal swabs are a reasonable alternative but may have tumor contamination due to monocyte infiltration.36
JMML-like neoplasms
There are defined JMML-like conditions that do not meet JMML diagnostic criteria. Noonan syndrome-associated myeloproliferative disorder (NAMD) is a distinct entity from JMML occurring in infants with Noonan syndrome and germline mutations in PTPN11, NRAS, KRAS, or RIT1.30 Clinically, NAMD may be milder, or may look just like JMML, including a blast percentage below 20% and hepatosplenomegaly. A subset of patients with a clinical presentation consistent with JMML but who lack a canonical RAS-pathway mutation will be found to harbor rearrangements in ALK, ROS1, or FLT3, and typically have monosomy 7 in addition to their fusion.37
Management of JMML and related disorders
The management of patients with JMML and JMML-like neoplasms varies based on clinical presentation, prognostic factors, and genetic drivers.35 Many children with JMML require hematopoietic stem cell transplant (HSCT), including those with PTPN11 and NF1 mutations or those with KRAS and NRAS mutations who have high-risk features including high DNA methylation.38 These patients are typically treated with 5-azacytidine with or without chemotherapy prior to HSCT. Patients with non-RAS- pathway mutations may receive targeted therapy (ie, ALK or FLT3 inhibitors) with or without chemotherapy followed by HSCT.37 Lower-risk patients who can be closely observed without intervention if they are clinically stable include patients with germline CBL mutations, patients with KRAS- and NRAS-mutant JMML who have low DNA methylation, and children with NAMD. For lower- risk patients who have visceral organ involvement leading to failure to thrive, respiratory distress, or diarrhea, treatment with oral 6-mercaptopurine or low-dose cytarabine as a temporizing measure is often indicated. Some JMML patients may be able to be treated with 5-azacytidine alone and avoid HSCT, but it is currently challenging to identify these patients at diagnosis.35
Differential considerations
Additional disorders that can mimic JMML or a JMML-like illness include certain viral or atypical infections (such as cytomegalovirus, human herpesvirus 6, mycobacteria, or toxoplasma).39 Of note, JMML often presents concomitantly with viral infections. Other rare etiologies include leukocyte adhesion deficiency and infantile malignant osteopetrosis.40 In the setting of peripheral blasts, it is important to make sure acute myeloid leukemia and chronic myeloid leukemia are ruled out. Some patients with KMT2A or NUP98 rearrangements or t(8;16)(p11;p13) may have features resembling JMML; due to clinical and biological differences, these disorders are classified as acute myeloid leukemia even when blasts are less than 20%.5,35,41,42
CASE 2 (Outcome)
The patient was treated with antibiotics resulting in resolution of the abscess and leukocytosis, but monocytosis persisted. Genetic testing revealed a germline heterozygous mutation in CBL and copy neutral loss of heterozygosity in the blood, thereby identifying a case of CBL-related JMML. Caused by germline mutations in the CBL gene, CBL syndrome has been identified as an underlying genetic predisposition to JMML.43 The clinical features of patients with CBL syndrome closely mirror those seen in classic Noonan syndrome, leading to the designation as a Noonan syndrome-like disorder.44 While CBL syndrome is caused by a heterozygous CBL mutation, the leukemia cells in patients with CBL syndrome harbor biallelic CBL mutations due to acquired uniparental disomy.45 JMML in patients with CBL syndrome usually follows a benign course, and patients may experience spontaneous disease regression; however, this must be differentiated from patients with somatic CBL mutations, who tend to present with more aggressive disease. Given that this patient was found to have a germline CBL mutation, he was monitored closely without intervention and was noted to have spontaneous resolution of all symptoms by 3 years of age.
Conclusion
While leukocytosis in young children may be due to various conditions, it is important to remember the unique myeloproliferative disorders of childhood. Clinicians must have a high index of suspicion for these disorders, as they can be aggressive and life-threatening. In infants and young children with myeloid- predominant leukocytosis, thorough evaluation to ensure timely identification of myeloproliferative disorders of childhood is essential for proper management and follow-up.
Acknowledgement
The authors wish to thank Irene Roberts and Kristie White for providing the peripheral blood smear images used.
Conflict-of-interest disclosure
Alexandra Satty: no competing financial interests to declare.
Elliot Stieglitz: no competing financial interests to declare.
Nicole Kucine: no competing financial interests to declare.
Off-label drug use
Alexandra Satty: discussion of off-label use of drugs is included.
Elliot Stieglitz: discussion of off-label use of drugs is included.
Nicole Kucine: discussion of off-label use of drugs is included.
