

Abstract
Thalassemia is an inherited red blood cell disorder whereby the qualitative and/or quantitative imbalance in α- to β-globin ratio results in hemolysis and ineffective erythropoiesis. Oxidative stress, from the precipitated excess globin and free iron, is a major factor that drives hemolysis and ineffective erythropoiesis. Pyruvate kinase activity and adenosine triphosphate availability are reduced due to the overwhelmed cellular antioxidant system from the excessive oxidative stress. Mitapivat, a pyruvate kinase activator in development as a treatment for thalassemia, was shown to increase hemoglobin and reduce hemolysis in a small phase 2 single-arm trial of patients with α- and β-thalassemia. The ongoing phase 3 studies with mitapivat and the phase 2 study with etavopivat will examine the role of pyruvate kinase activators as disease modifying agents in thalassemia.
Learning Objectives
Understand thalassemia as an inherited red blood cell disorder where both oxidative stress and hemolysis play a major role in the pathogenesis
Describe the results of the phase 2 single-arm trial of pyruvate kinase activator mitapivat in patients with α- and β-thalassemia
Appreciate that pyruvate kinase activation is the first mechanism targeted in clinical trials for patients with α-thalassemia
CLINICAL CASE
A 34-year-old female with non-deletional Hemoglobin H disease (α−3.7/Hemoglobin Constant Spring compound heterozygote) is referred by another hematologist for a second opinion regarding splenectomy. Patient receives on average 2-3 units of packed red blood cells per year for episodes of symptomatic anemia and has a liver iron concentration of 3.0 mg/g dw. She was advised to undergo splenectomy by the referring hematologist in hopes of improving her hemoglobin (Hb) and her symptoms, but the patient is concerned about the risk of infection and thrombosis associated with asplenia and wishes to explore alternatives to splenectomy. Patient on review has fatigue, exertional dyspnea, and exercise limitations. She comments on being frustrated by her profound fatigue and the need to take a nap almost daily. Other secondary causes of fatigue including depression have been ruled out. On examination, the patient has scleral icterus and palpable splenomegaly at 5 cm below the costal margin. Laboratory findings included a hemoglobin of 7.0 g/dL, MCV 65 fL, mean corpuscular hemoglobin 21 pg/ cell, absolute reticulocyte count 336 × 109/L, lactate dehydrogenase level 462 U/L, total bilirubin 3 mg/dL (indirect bilirubin 2.8 mg/dL), haptoglobin undetectable. Abdominal ultrasound demonstrated a spleen size of 18 cm in craniocaudal length.
Introduction
Thalassemia is an inherited red blood cell (RBC) disorder whereby the qualitative and/or quantitative imbalance in α- to β-globin ratio results in ineffective erythropoiesis and hemolysis.1 A paucity or qualitative deficiency in β-globin results in β-thalassemia and vice versa. Presentation is highly variable, ranging from those who do not require regular transfusions to survive (non-transfusion-dependent thalassemia [NTDT]) to severely anemic patients who are transfusion-dependent (TDT). NTDT is a highly prevalent condition in many areas around the globe and is becoming more prevalent in others due to migration. There is growing evidence that patients with NTDT face a high burden of morbidity and mortality despite not being transfusion dependent.1,2 Ineffective erythropoiesis and hemolysis drives anemia, hypercoagulability, and primary iron overload, resulting in a myriad of complications including gallstones, hepatosplenomegaly, bone deformities, osteoporosis, extramedullary hematopoiesis (EMH), leg ulcers, thrombosis, pulmonary hypertension, endocrinopathies, growth delay, liver fibrosis and cirrhosis, heart failure, and arrhythmia, among others (Figure 1).1 Treatment for NTDT is currently confined to treatment of these complications (e.g., hypertransfusion for EMH) since there is no approved disease-modifying therapy, and in particular, for patients with α-thalassaemia. Likewise, treatment for TDT is also largely confined to regular transfusions and supportive care. The ensuing iron overload from regular transfusions further exacerbates the liver, cardiac, endocrine, and renal complications (Figure 1).1 Use of splenectomy for both NTDT and TDT are in decline because of the risk of infection and thrombosis.1 While luspatercept is currently approved for β-TDT, it reduces the transfusion burden by one-third or more (and at least 2 units over 12 weeks) from baseline in only 21.4% of patients and must be administered as subcutaneous injections every 3 weeks.3 Gene therapy is approved for use in a small segment of patients with β-TDT thalassaemia. Since hemolysis plays a crucial role in the pathogenesis of thalassemia, agents that can combat the hemolysis would be desirable. To this end, two compounds, mitapivat and etavopivat, have been developed and are currently undergoing clinical trials in patients with thalassemia. Mitapivat and etavopivat are two oral small molecular allosteric activators of the RBC pyruvate kinase (PKR), the last enzyme in glycolysis that converts phosphoenolpyruvate to pyruvate. Mitapivat is approved by the FDA for treatment of pyruvate kinase deficiency, and both mitapivat and etavopivat are currently evaluated for the treatment of thalassemia and sickle cell disease (SCD). RBCs are entirely dependent on glycolysis (due to the lack of mitochondria) to produce energy (e.g., adenosine triphosphate [ATP]), needed for the maintenance of cellular metabolism, hydration, membrane integrity, and deformability, and to provide antioxidant capacity to protect hemoglobin molecules and other proteins from continuous oxidative stress.4 Any mismatch in energy (ATP) supply and demand leads to reduced RBC health and lifespan. How this mismatch occurs in thalassemia and can be attenuated by pyruvate kinase (PK) activation will be detailed below.
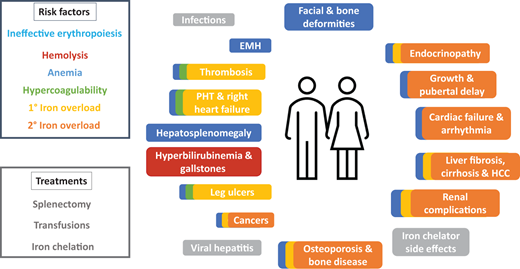
Multisystem complications from risk factors and treatments of thalassemia. Risk factors and their corresponding complications are color-coded. Complications with overlapping colors denote those stemming from and exacerbated by the multiple risk factors involved. Modified from Taher AT, Musallam KM, Cappellini MD. β-Thalassemias. N Engl J Med. 2021;384(8):727-743. 1°, primary; 2°, secondary; EMH, extramedullary hematopoiesis; HCC, hepatocellular carcinoma; PHT, pulmonary hypertension.
Multisystem complications from risk factors and treatments of thalassemia. Risk factors and their corresponding complications are color-coded. Complications with overlapping colors denote those stemming from and exacerbated by the multiple risk factors involved. Modified from Taher AT, Musallam KM, Cappellini MD. β-Thalassemias. N Engl J Med. 2021;384(8):727-743. 1°, primary; 2°, secondary; EMH, extramedullary hematopoiesis; HCC, hepatocellular carcinoma; PHT, pulmonary hypertension.
Ineffective erythropoiesis and hemolysis in β-thalassemia
In β-thalassemia, excess α-globin overwhelms the α hemoglobin stabilizing protein, which normally stabilizes the free α-globin chains. The free α-globin aggregates and precipitates, forming inclusion bodies called Fessas bodies.5 In normal erythropoiesis, GATA1 promotes terminal erythroid differentiation, and GATA1 is protected by Heat Shock Protein 70 (HSP70) in the nucleus. In β-thalassemia, the free α-globin sequesters HSP70 in the cytoplasm, aided by exportin-1 (Figure 2).6 HSP70, having been lured away, is unable to protect GATA1 from degradation by Caspase 3, leading to accumulation and apoptosis of the erythroid precursors in the bone marrow, termed ineffective erythropoiesis.7 Moreover, free α-globin molecules auto-oxidize and form hemichromes (α-globin with oxidized ferric iron) and reactive oxygen species (ROS). The hemichromes precipitate and intercalate into the RBC plasma membrane and oxidize the membrane lipid and proteins.7 The resultant destabilization of the RBC membrane cannot be readily detoxified by the cellular antioxidant system, and the excess ROS leads to intramedullary cell death. If by chance the RBCs survive the maturation journey, these cells have diminished lifespan due to the oxidative stress, and hemolysis ensues in the peripheral circulation (Figure 2). Oxidative injury causes Band 3 to cluster, producing a neoantigen that binds to the immunoglobulin G and complement, resulting in removal of the damaged RBC by macrophages in the circulation. In addition, the free iron liberated from the hemolysis creates highly reactive hydroxyl and hydroperoxyl radicals, which exerts further oxidative stress and tissue damage.7
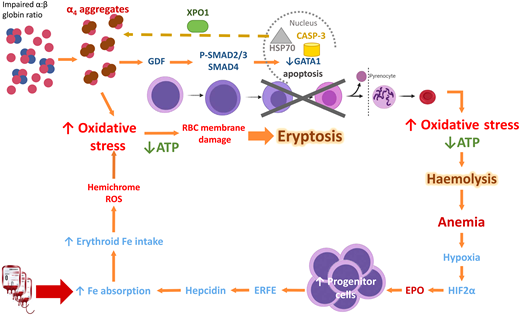
Mechanism of oxidative stress, ineffective erythropoiesis and hemolysis in β-thalassemia. Impaired α- to β-globin ratio results in aggregation and precipitation of the excess α-globin, exerting oxidative stress on the developing RBC. Concurrently, α-globin aggregates induce the sequestration of HSP70 in the cytoplasm, GDF activation and phosphorylation of SMAD2/3, and activation of SMAD4. Both processes lead to reduced GATA1 resulting in eryptosis. RBCs that exit the marrow soon undergo hemolysis because of shortened RBC lifespan. Modified from Longo F, Piolatto A, Ferrero GB, Piga A. Int. J. Mol. Sci. 2021, 22, 7229, Taher A and Saliba AN. Hematology Am Soc Hematol Educ Program. 2017 Dec 8;2017(1):265-271; Musallam KM, et al. Haematologica. 2011;96(11):1605-1612; Rivella S. Haematologica. 2015;100(4):418. ATP, adenosine triphosphate; EPO, erythropoietin; ERFE, erythroferrone; GDF, growth differentiation factor; HIF2α, hypoxic inducible factor 2α; HSP70, Heat Shock Protein 70; RBC, red blood cell; ROS, reactive oxygen species; XPO-1, exportin-1.
Mechanism of oxidative stress, ineffective erythropoiesis and hemolysis in β-thalassemia. Impaired α- to β-globin ratio results in aggregation and precipitation of the excess α-globin, exerting oxidative stress on the developing RBC. Concurrently, α-globin aggregates induce the sequestration of HSP70 in the cytoplasm, GDF activation and phosphorylation of SMAD2/3, and activation of SMAD4. Both processes lead to reduced GATA1 resulting in eryptosis. RBCs that exit the marrow soon undergo hemolysis because of shortened RBC lifespan. Modified from Longo F, Piolatto A, Ferrero GB, Piga A. Int. J. Mol. Sci. 2021, 22, 7229, Taher A and Saliba AN. Hematology Am Soc Hematol Educ Program. 2017 Dec 8;2017(1):265-271; Musallam KM, et al. Haematologica. 2011;96(11):1605-1612; Rivella S. Haematologica. 2015;100(4):418. ATP, adenosine triphosphate; EPO, erythropoietin; ERFE, erythroferrone; GDF, growth differentiation factor; HIF2α, hypoxic inducible factor 2α; HSP70, Heat Shock Protein 70; RBC, red blood cell; ROS, reactive oxygen species; XPO-1, exportin-1.
Oxidative stress and hemolysis in α-thalassemia
Conversely, in α-thalassemia, excess β-globin and γ-globin form tetramers called hemoglobin H and Barts, respectively. Although hemoglobin H (HbH) and Barts are more stable than α globin tetramers, they are still liable to induce the ROS formation and cause damage to the developing RBC. The phenotype of HbH disease, a form of α-thalassemia where three of the four α- globin genes are mutated, is characterized by chronic hemolytic anemia.8 The exact mechanism by which α-thalassemia leads to hemolysis and ineffective erythropoiesis is less clear, but abundant evidence suggests that the process may be analogous to β-thalassemia in that oxidative stress plays an important component in the pathophysiology. This includes findings of low glutathione levels,9 hepcidin, total antioxidant capacity levels,10 and elevated malondialdehyde levels in patients with α-thalassemia.11
Impairment of glycolysis in thalassemia
There is also evidence to show that in thalassemia, the glycolytic pathway is affected by the oxidative stress. Ting et al determined, via 13 C and 31P magnetic resonance spectroscopy, that glucose metabolism in the RBCs of patients with β-thalassemia intermedia were significantly (approximately three times) higher than in healthy patients or patients with β-thalassemia trait, even when contribution by reticulocytes were excluded.12 However, ATP concentrations in the RBCs of patients with β-thalassemia major or Hemoglobin E/β-thalassemia were significantly lower than healthy controls.13 These two studies together show that glucose metabolism is shifted away from the glycolytic pathway and toward the pentose phosphate pathway. PK enzyme activity and stability were found to be reduced in patients with TDT with no pathogenic PKLR mutation, even when adjusted for RBC age and the presence of reticulocytosis.14 Matte et al have shown that ROS levels were increased in the RBCs of Hbbth3/+ mice compared to wild-type mice, and that PKR and PKM2 expression was higher in both the reticulocyte and older RBC fractions.15 PKM2 was also upregulated in the RBCs of Hbbth3/+ mice and may act as a compensatory response to the increased oxidative stress.15 These discoveries led to the hypothesis that pyruvate kinase activation may reduce oxidative damage, thus reducing hemolysis, improving ineffective erythropoiesis and RBC lifespan, and resulting in improvement of anemia. Also, improvement in ineffective erythropoiesis may alleviate the dysregulated iron metabolism, leading to reduction in iron overload (Figure 3).
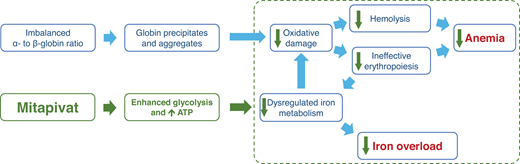
Hypothesis on how pyruvate kinase activation, through enhanced glycolysis and increased in ATP availability, reduces oxidative damage and hemolysis and improves ineffective erythropoiesis and dysregulated iron metabolism, thereby improving anemia and iron overload.
Hypothesis on how pyruvate kinase activation, through enhanced glycolysis and increased in ATP availability, reduces oxidative damage and hemolysis and improves ineffective erythropoiesis and dysregulated iron metabolism, thereby improving anemia and iron overload.
Pyruvate kinase activation in thalassemia mouse model
To examine the effect of pyruvate kinase activation in thalassemia, Matte et al administered mitapivat to Hbbth3/+ mice at 50 mg/kg twice daily for 21 days (by gavage) and 56 days (by oral diet), respectively. After 21 days, improvements were observed in Hb, mean corpuscular volume (MCV), mean corpuscular Hb, RBC morphology, and survival (14 vs 9.6 days in treated and untreated Hbbth3/+ mice compared to 18.9 days in wild-type mice). Concomitant reduction in markers of hemolysis (lactate dehydrogenase [LDH], total and indirect bilirubin), erythropoietin (EPO) level, reticulocyte count, ROS, hemichromes, and α-globin membrane precipitates was observed, accompanied by an increased glutathione to glutathione disulfide ratio. An increase in ATP was also observed. In addition to the amelioration of hemolysis, mitapivat improved ineffective erythropoiesis. Splenic extramedullary hematopoiesis was reduced, and PK activity was increased in RBC polychromatic and orthochromatic erythroblasts of mitapivat-treated Hbbth3/+ mice, accompanied by reduced apoptotic (annexin-V+) erythroblasts and ROS in maturing erythroblasts.15 This also resulted in a reduction in the erythroferrone (ERFE) level, followed by upregulation of liver hepcidin expression and a reduction in hepatic iron overload. There is evidence that mitapivat may also reduce duodenal iron absorption via the PKM2-HIF2α axis by downregulation of HIF2α, NF-κb p65 active form, FPN1, and Dmt1 in the enterocytes of the mitapivat-treated Hbbth3/+ mice.15
Recently, Mattè et al demonstrated that in chronically transfused Hbbth3/+ mice treated with mitapivat, the results were a longer interval between transfusions (13.8 vs 10.5 days in mitapivat and vehicle-treated mice, respectively) and reduced splenic iron accumulation.16 Similar to the Hbbth3/+ mice not on transfusion treated with mitapivat, α-globin membrane precipitates, EPO level and liver iron accumulation were reduced, hepcidin increased, and splenic macrophage function was shifted from a pro-inflammatory to a pro-resolving state.16 Concomitant administration of mitapivat with deferiprone had a similar effect.16 These findings provided the preclinical evidence for subsequent trials of pyruvate kinase activation in both TDT and NTDT.
CLINICAL CASE (continued)
The patient was once again counseled on the benefits and risks of splenectomy, including the potential benefit of increasing total hemoglobin by 1-2 g/dL, but she declined this approach based on the four- to five-fold risk of venous thromboembolism and pulmonary hypertension compared to non-spelenectomized patients.17 Regular transfusion was also offered as an option, but the patient was concerned about the need for chronic iron chelation. Given the patient's hemolytic profile, she was offered the opportunity to participate in a clinical trial of a pyruvate kinase activator.
Pyruvate kinase activation in thalassemia patients
Mitapivat
The phase 2, open-label, multicenter study investigated the efficacy and safety of mitapivat in adult patients with α- and β-NTDT.18 The primary objective of the study was to evaluate the efficacy of mitapivat in NTDT. Twenty patients with NTDT from four sites across the US, UK, and Canada were enrolled. The genotype inclusion criterion was broad and included β-thalassemia with or without α-globin gene mutations, HbE/β-thalassemia, or α-thalassemia. Patients had to have a hemoglobin less than 10 g/dL, and non-transfusion dependency was defined as less than 6 RBC units transfused in the preceding 24 weeks and none in the 8 weeks prior to study drug-dosing. Following screening, patients entered a 24-week core period where they received an initial dose of mitapivat 50 mg twice a day orally. At week 6, the dose was increased to 100 mg twice a day based on safety and tolerability. After the 24-week core period, patients may enter an optional 10-year extension period. The primary endpoint was defined as an increase of hemoglobin by at least 1 g/dL from baseline between weeks 4 and 12. Secondary and exploratory endpoints included sustained or delayed Hb response, markers of hemolysis, hematopoietic activity, and safety. The secondary endpoint was defined as meeting the primary response and at least a 1 g/dL increase in hemoglobin in at least two readings between weeks 12 and 24. The median age in this cohort was 44 years, 50% of the population was Asian, and the median Hb at baseline was 8.4 g/dL.18 Overall, 5 patients had α-thalassemia (HbH), and 15 patients had β-thalassemia. The primary endpoint was met in 80% (16/20) of the participants; all 5 patients with α-thalassemia and 11/15 patients with β-thalassemia met the primary endpoint. The secondary endpoint of sustained hemoglobin response was met in 65% of the 20 patients, with all 5 patients with α-thalassemia meeting this endpoint. The mean change in Hb from baseline over a 12-week interval between weeks 12 and 24 was similar between α- and β-thalassemia (1.2 and 1.3 g/dL, respectively), and the mean time to ≥1 g/dL increase in Hb among the responders was 4.5 weeks.18 Qualitatively, markers of hemolysis (LDH and bilirubin) and erythropoiesis were reduced or remained stable in most patients. Consistent with the mechanism of action of mitapivat, the mean ATP change in blood ranged from 62-87%, similar to what was previously observed in a multiple-ascending dose study in healthy volunteers (60% maximum increase in ATP).18 Note that all participants in this cohort had nonpathogenic PKLR genotypes.
Safety of mitapivat in thalassemia
In terms of safety, 17 (85%) patients experienced a treatment-emergent adverse event during the 24-week core period, with most being grade 1 or 2; the majority were self-limiting. The most commonly reported adverse events were initial insomnia (n = 10 [50%]), dizziness (n = 6 [30%]), and headache (n = 5 [25%]).18 The safety profile was similar to prior studies of mitapivat in healthy volunteers and patients with PK deficiency. In the 17 participants who enrolled in the 10-year long-term extension (LTE), Hb increase was sustained with a mean increase in Hb of 1.7 g/dL. Qualitatively, continued reduction in LDH, indirect bilirubin, and EPO was observed in the LTE up to week 72, as well as a reduction (expressed as median change from baseline) in ERFE (−5430 ng/L), reticulocytes/erythrocyte ratio (−0.007), and soluble transferrin receptor level (−1.82 mg/L), while hepcidin remained stable over time (+650 ng/L).19 The type and frequency of adverse events in the extension period were consistent with those observed in the core period. Initial insomnia was confined to the core period and was not observed in the LTE. Adverse events occurring in ≥15% of patients during the extension period were headache (5/17) and back pain (3/17), none of which were grade ≥3. In patients who have had repeat bone mineral density in the extension period, no trends for decreases were observed.
Phase 3 trial of mitapivat in thalassemia
The encouraging results from the phase 2 study have led to the development of two phase 3, multicenter, randomized, double- blind, placebo-controlled studies of mitapivat, in patients with non-transfusion dependent (ENERGIZE; NCT04770753) and transfusion-dependent α- or β-thalassemia (ENERGIZE-T; NCT04770779) (Table 1).20 In ENERGIZE, 171 adults with either α- or β-thalassemia (based on Hb electrophoresis, Hb HPLC, and/or DNA analysis), a Hb ≤10 g/dL, and non-transfusion dependency (defined as ≤5 RBC units during the 24-weeks before randomization and no RBC transfusions within the 8 weeks prior) are enrolled in the 24-week double-blind core period and randomized in a 2:1 fashion to receive either mitapivat 100 mg or placebo twice daily. The primary endpoint is Hb response, its definition being similar to the primary endpoint in the phase 2 study. However, the secondary endpoints include a patient-reported outcome and functional assessment (change in Functional Assessment of Chronic Illness Therapy [FACIT]-Fatigue subscale score, patient-reported global measures of fatigue, 6-minute walk test distance) in addition to change in average Hb concentration, markers of hemolysis, erythropoiesis, and iron metabolism. The design of ENERGIZE-T is similar, with key differences being a 48-week double-blind core period enrolling 240 adult α- or β-TDT (e.g., patients who are transfusion-dependent with HbH disease or α-thalassemia major/Hb Barts hydrops fetalis would qualify), the primary endpoint being transfusion reduction response (defined as a 50% or greater reduction in transfused RBC units with a reduction of two or more units of transfused RBCs in any consecutive 12-week period through week 48 compared with baseline), and the secondary endpoints examining transfusion reduction at different degrees and intervals within the 48-week core period. Transfusion-dependency is defined as 6 to 20 RBC units transfused and no transfusion-free period for more than 6 weeks during the 24-weeks before randomization. In both studies, eligible patients have the option of entering the 5-year open-label extension. Both studies also permit the enrollment of patients on stable hydroxyurea dose for at least 16 weeks before randomization, recognizing that it is used by some centers in the treatment of patients with β-thalassemia.17
Clinical trials of pyruvate kinase activators in thalassemia
| Category | Phase | Drug | N | Duration | Identifier | Status |
|---|---|---|---|---|---|---|
| NTDT | 2 (single-arm, open-label) | Mitapivat | 20 | 24 weeks | NCT03692052 | Completed |
| Etavopivat | 20 | 48 weeks | NCT04987489 (cohort C) | Ongoing | ||
| 3 (double-blind RCT) | Mitapivat: placebo (2:1) | 171 | 24 weeks | NCT04770753 | Ongoing | |
| TDT | 2 (single-arm, open-label) | Etavopivat | 20 | 48 weeks | NCT04987489 (cohort B) | Ongoing |
| 3 (double-blind RCT) | Mitapivat: placebo (2:1) | 240 | 48 weeks | NCT04770779 | Ongoing |
| Category | Phase | Drug | N | Duration | Identifier | Status |
|---|---|---|---|---|---|---|
| NTDT | 2 (single-arm, open-label) | Mitapivat | 20 | 24 weeks | NCT03692052 | Completed |
| Etavopivat | 20 | 48 weeks | NCT04987489 (cohort C) | Ongoing | ||
| 3 (double-blind RCT) | Mitapivat: placebo (2:1) | 171 | 24 weeks | NCT04770753 | Ongoing | |
| TDT | 2 (single-arm, open-label) | Etavopivat | 20 | 48 weeks | NCT04987489 (cohort B) | Ongoing |
| 3 (double-blind RCT) | Mitapivat: placebo (2:1) | 240 | 48 weeks | NCT04770779 | Ongoing |
RCT, randomized controlled trial.
Etavopivat
Etavopivat is also being evaluated in NTDT and TDT following the conclusion of the single-ascending dose and multiple-ascending dose phase 1 trial (NCT03815695) in 90 healthy adults.21 The ongoing phase 2 open-label study (NCT04987489) aims to evaluate the safety and efficacy of etavopivat in patients 12 to 65 years old with SCD on chronic transfusions (cohort A), TDT (cohort B), and NTDT (cohort C) (Table 1).22 Patients will receive etavopivat 400 mg once daily for 48 weeks for all cohorts. The primary endpoint for the TDT cohort is erythroid response, defined as the proportion of patients with 20% or greater reduction in transfusion over a continuous 12-week treatment period compared to baseline. The primary endpoint for the NTDT cohort is defined as a 1.0 g/dL or greater increase from baseline at week 12 in Hb.22 Both the TDT and NTDT cohorts plan to enroll 12 to 20 patients and are powered to detect a response rate of 60% and 50% to the primary endpoint in the TDT and NTDT cohorts, respectively. Secondary and exploratory endpoints include other transfusion reduction and hemoglobin response parameters, changes in HRQoL (SF-36 and PROMIS), serum ferritin levels, liver iron, 2,3-BPG, ATP, PK, and safety parameters.22
CLINICAL CASE (continued)
The patient was enrolled in the phase 2 study of mitapivat in NTDT. Her hemoglobin increased by 1.0 g/dL from baseline within 4 weeks of starting treatment and reached 1.4 g/dL at 24 weeks. She had initial insomnia, which abated by day 5 on treatment without intervention. Her markers of hemolysis improved and EPO level reduced. Subjectively, the patient noted that her exercise capacity increased, and most important to her, she no longer needed to take a nap in the afternoon.
Considerations on the role of pyruvate kinase activation in the treatment of thalassemia
The preclinical and clinical evidence for pyruvate kinase activation in thalassemia and other hemolytic disorders like pyruvate kinase deficiency and SCD thus far points to its effectiveness at ameliorating hemolysis brought on by oxidative stress. As such, one may surmise that it may be more effective in thalassemia genotypes that result in a predominantly hemolytic phenotype (e.g., non-β0/β0 thalassemia or HbH disease). However, no distinct relationship between genotype and hemoglobin response could be gleaned from the phase 2 data of mitapivat in thalassemia because 80% of patients met the primary endpoint, despite their diverse thalassemia genotype, as noted by Kuo et al.18 The fact that mitapivat also improved ineffective erythropoiesis in Hbbth3/+ mice suggests that mitapivat may have comparable efficacy in patients whose pathophysiology is dominated by ineffective erythropoiesis. The ongoing phase 2 and 3 trials may provide further insight into how pyruvate kinase activation may be effectively used in the treatment of patients with thalassemia.
Conclusion
Despite advances in the understanding in the pathophysiology of thalassemia, treatment of patients with NTDT, and in particular α-thalassemia, remains supportive. Luspatercept is the only approved treatment for patients with TDT aside from transfusion. Pyruvate kinase activation was able to increase hemoglobin concentration and reduce hemolysis in the majority of the patients with thalassemia in the phase 2 study, despite their diverse genotypic heterogeneity, which provides the proof of concept that suppression of hemolysis may improve the pathology of both α- and β-thalassemia. The ongoing phase 3 trials of mitapivat and the phase 2 trials of etavopivat in α- and β-thalassemia will determine whether pyruvate kinase activation will improve patient-important outcomes by assessing their functional and quality-of-life impact beyond change in hemoglobin and markers of hemolysis and erythropoiesis.
Conflict-of-interest disclosure
Kevin H.M. Kuo: consultancy and research funding: Agios Pharmaceuticals, Pfizer; consultancy: Alexion Pharmaceuticals, NovoNordisk, Vertex Pharmaceuticals; consultancy and honoraria: Bristol Myers Squibb; data safety monitoring board: Bioverativ/Sanofi/Sangamo.
Off-label drug use
Kevin H.M. Kuo: nothing to disclose.
