

Key Points
CatG cleavage of PAR4 generates a novel tethered ligand that induces PAR4 activation and platelet signaling and aggregation.
Elucidation of the molecular basis of CatG cleavage of human PAR4 expands our understanding of protease activation of PARs.

Abstract
Platelet-neutrophil interactions regulate ischemic vascular injury. Platelets are activated by serine proteases that cleave protease-activated receptor (PAR) amino termini, resulting in an activating tethered ligand. Neutrophils release cathepsin G (CatG) at sites of injury and inflammation, which activates PAR4 but not PAR1, although the molecular mechanism of CatG-induced PAR4 activation is unknown. We show that blockade of the canonical PAR4 thrombin cleavage site did not alter CatG-induced platelet aggregation, suggesting CatG cleaves a different site than thrombin. Mass spectrometry analysis using PAR4 N-terminus peptides revealed CatG cleavage at Ser67-Arg68. A synthetic peptide, RALLLGWVPTR, representing the tethered ligand resulting from CatG proteolyzed PAR4, induced PAR4-dependent calcium flux and greater platelet aggregation than the thrombin-generated GYPGQV peptide. Mutating PAR4 Ser67or Arg68 reduced CatG-induced calcium flux without affecting thrombin-induced calcium flux. Dog platelets, which contain a conserved CatG PAR4 Ser-Arg cleavage site, aggregated in response to human CatG and RALLLGWVPTR, while mouse (Ser-Gln) and rat (Ser-Glu) platelets were unresponsive. Thus, CatG amputates the PAR4 thrombin cleavage site by cleavage at Ser67-Arg68 and activates PAR4 by generating a new functional tethered ligand. These findings support PAR4 as an important CatG signaling receptor and suggest a novel therapeutic approach for blocking platelet-neutrophil-mediated pathophysiologies.
Introduction
Human platelets express 2 G-protein coupled, protease-activated receptors (PAR1 and PAR4).1 Although many serine proteases activate platelets through PARs,2 thrombin is the best studied. PARs are activated by proteolysis of their amino termini.1 The resulting new N-terminus functions as a tethered ligand that binds to an extracellular loop of the PAR,3,4 resulting in platelet activation. Compared with PAR1, PAR4 has been less studied, but recent findings emphasize its importance and potential as a therapeutic target.2 PAR1 has a higher affinity for thrombin, and Ca2+ transients rise and fall sharply after activation. In contrast, PAR4 acts as an “amplifier,” inducing a gradual but sustained rise in Ca2+ that accounts for greater intracellular Ca2+ flux, thrombin generation, and fibrin formation under shear stress.5,6
Elevated blood neutrophil levels correlate with ischemic arterial events,7 and platelet-leukocyte aggregates are elevated during a stroke.8 Activated neutrophils release the serine protease cathepsin G (CatG) at sites of injury and inflammation.9 CatG is a potent human PAR4-dependent platelet agonist,10-13 but it is unknown where CatG cleaves PAR4 and how this cleavage might activate the receptor.
Methods
A more detailed methods section is provided in the supplemental Data.
Platelet and neutrophil isolation
PAR4 calcium flux assays
PAR4-dependent calcium flux was measured using Fura2-QBT in platelets and HEK293T/17 cells with wild-type (WT) and mutant PAR4.16
PAR-4 peptide and cleavage analysis
PAR4 synthetic peptides were incubated with protease, snapped frozen, and sent for mass spectrometry analysis.
Results and discussion
CatG elicits platelet activation and aggregation through PAR4 signaling
Of the major serine proteases released from neutrophils, CatG was the only protease to induce platelet aggregation and αIIbβ3 activation (supplemental Figure 1A-D). Supplemental Figure 1A shows the effect of neutrophil activation on platelet activation is mediated in large part by CatG. Using PAR-specific inhibitors, we observed CatG-dependent platelet activation was mediated by PAR4 and not PAR1 (Figure 1A; supplemental Figure 2). Because platelet-neutrophil interactions are critical in stroke pathophysiology and a common PAR4 Ala120Thr variant has been associated with human stroke risk,17,18 the effect of this variant on CatG-induced platelet activation was studied. Compared with platelets from homozygous Ala120 individuals, CatG stimulation of platelets homozygous for the racially divergent and hyperreactive PAR4 Thr120 variant demonstrated modest but significantly greater αIIbβ3 activation and P-selectin expression (Figure 1B; supplemental Figure 3). Thus, neutrophil-released CatG may contribute to the increased stroke risk associated with the PAR4Thr120 variant.
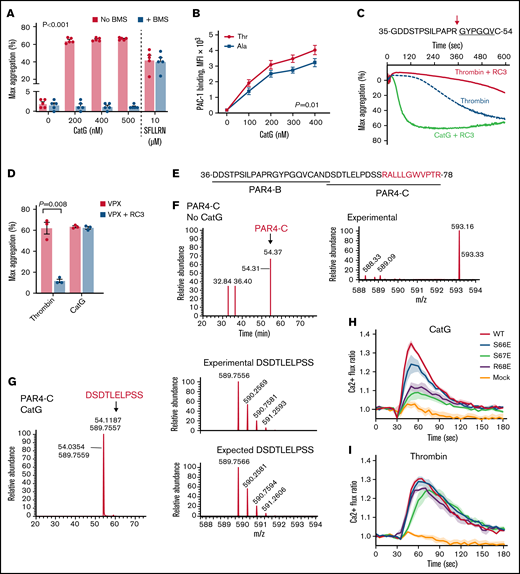
Neutrophil cathepsin G cleaves PAR4 at Ser67-Arg68 to induce platelet aggregation. (A) Washed platelets were treated with varying concentrations of CatG in the presence or absence of the PAR4 inhibitor, BMS-986120 (400 nM BMS; n = 5), and maximum(max)platelet aggregation (percent ± SEM) recorded. The PAR1 activation peptide (SFLLRN; 10 μM) served as a negative control (n = 5) for PAR4 inhibition. (B) Washed platelets were stimulated with increasing concentrations of CatG, and platelet activation was measured by PAC-1 binding (Ala: n = 7; Thr: n = 7) and displayed as MFI (mean ± SEM). (C) PAR4 amino acid sequence targeted by RC3 monoclonal antibody shown above. The underline represents the tethered ligand generated by thrombin. The arrow represents the location of the canonical thrombin cleavage site. All aggregation studies were performed with PAR1 blockade using 100 nM vorapaxar. Representative tracing of washed platelets treated with 0.25 U/mL thrombin or 200 nM CatG in the presence or absence of RC3. (D) Quantification of maximum (max) platelet aggregation (mean percent ± SEM) (n = 3 different subjects). (E) PAR4-B and PAR4-C peptide sequences used in CatG proteolysis analysis by LC-MS/MS. Red sequence indicates novel tethered ligand generated by CatG. (F) LC-MS/MS performed on PAR4-C in the absence of CatG. Time of flight analysis showed an experimental peak with the correct mass (m) over charge (z) ratio. (G) LC-MS/MS analysis performed on PAR4-C after incubation with 400 nM CatG at 37°C for 15 minutes. Time of flight analysis observed a peak with the correct m/z ratio of a fragment containing the amino acids DSDTLELPSS (the last residue is Ser67), indicating CatG cleaved PAR4-C between Ser67 and Arg68. For reference, the expected m/z ratio of DSDTLELPSS is shown below. (H) Calcium mobilization of WT, mutated PAR4, or empty vector (mock) expressed in HEK293T/17 cells treated with or without CatG (2.5 µM) in the presence of PAR1 blockade with 100 nM vorapaxar. (I) WT, mutated PAR4, or empty vector (mock) were expressed in HEK293T/17 cells and treated with thrombin (1.5 U/mL) in the presence of 100 nM vorapaxar. Solid thick lines and thin vertical lines are means and SEMs, respectively. n = 4 independent experiments performed in duplicate in panels H-I.
Neutrophil cathepsin G cleaves PAR4 at Ser67-Arg68 to induce platelet aggregation. (A) Washed platelets were treated with varying concentrations of CatG in the presence or absence of the PAR4 inhibitor, BMS-986120 (400 nM BMS; n = 5), and maximum(max)platelet aggregation (percent ± SEM) recorded. The PAR1 activation peptide (SFLLRN; 10 μM) served as a negative control (n = 5) for PAR4 inhibition. (B) Washed platelets were stimulated with increasing concentrations of CatG, and platelet activation was measured by PAC-1 binding (Ala: n = 7; Thr: n = 7) and displayed as MFI (mean ± SEM). (C) PAR4 amino acid sequence targeted by RC3 monoclonal antibody shown above. The underline represents the tethered ligand generated by thrombin. The arrow represents the location of the canonical thrombin cleavage site. All aggregation studies were performed with PAR1 blockade using 100 nM vorapaxar. Representative tracing of washed platelets treated with 0.25 U/mL thrombin or 200 nM CatG in the presence or absence of RC3. (D) Quantification of maximum (max) platelet aggregation (mean percent ± SEM) (n = 3 different subjects). (E) PAR4-B and PAR4-C peptide sequences used in CatG proteolysis analysis by LC-MS/MS. Red sequence indicates novel tethered ligand generated by CatG. (F) LC-MS/MS performed on PAR4-C in the absence of CatG. Time of flight analysis showed an experimental peak with the correct mass (m) over charge (z) ratio. (G) LC-MS/MS analysis performed on PAR4-C after incubation with 400 nM CatG at 37°C for 15 minutes. Time of flight analysis observed a peak with the correct m/z ratio of a fragment containing the amino acids DSDTLELPSS (the last residue is Ser67), indicating CatG cleaved PAR4-C between Ser67 and Arg68. For reference, the expected m/z ratio of DSDTLELPSS is shown below. (H) Calcium mobilization of WT, mutated PAR4, or empty vector (mock) expressed in HEK293T/17 cells treated with or without CatG (2.5 µM) in the presence of PAR1 blockade with 100 nM vorapaxar. (I) WT, mutated PAR4, or empty vector (mock) were expressed in HEK293T/17 cells and treated with thrombin (1.5 U/mL) in the presence of 100 nM vorapaxar. Solid thick lines and thin vertical lines are means and SEMs, respectively. n = 4 independent experiments performed in duplicate in panels H-I.
Thrombin cleavage of PAR4 at Arg47-Gly48 is blocked by the monoclonal antibody RC3.6 RC3 had little effect on CatG-induced platelet aggregation despite largely abolishing thrombin-induced aggregation (Figure 1C-D; supplemental Figure 4) (note thrombin activation of PAR1 was blocked in Figure 1C-D, as determined by supplemental Figure 4). These results suggest CatG cleaves PAR4 at a site different than thrombin. It is not clear why our data apparently differ from previous observations using a polyclonal antibody to PAR46,13 since the antibodies were raised against the same sequence, but perhaps the polyclonal antibody also blocked the CatG cleavage site while monoclonal RC3 does not.
CatG proteolysis of PAR4 N-terminal extracellular sequence
To examine the possible cleavage fragments generated by CatG, 2 portions of the PAR4 extracellular N-terminus were synthesized and analyzed by LC-MS/MS: PAR4-B containing the thrombin cleavage site, and PAR4-C, downstream of the thrombin cleavage site (Figure 1E). As expected, PAR4-B was cleaved by thrombin between amino acids Arg47 and Gly48, while little cleavage was observed with CatG (supplemental Figure 5). In contrast, thrombin failed to cleave PAR4-C, while CatG cleavage generated several fragments, including DSDTLELPSS (Figure 1G; and data not shown). Identification of the DSDTLELPSS fragment (amino-terminal sequence of PAR4-C) indicated CatG cleavage between Ser67 (S67)-Arg68 (R68). As a control, in the absence of thrombin or CatG, PAR4-B and PAR4-C remained intact in these analyses (Figure 1F; supplemental Figure 5).
Mutations of amino acids Ser67 and Arg68 decrease CatG-stimulated PAR4-mediated calcium signaling
To specifically examine if CatG induces PAR4 activation and signaling by cleaving Ser67-Arg68, we assessed calcium flux using various PAR4 mutants. HEK cells transfected with WT PAR4 demonstrated increased calcium flux when treated with CatG compared with mock transfection (Figure 1H). CatG activation of HEK cells expressing either PAR4 mutated Ser67 to Glu (S67E) or Arg68 to Glu (R68E) resulted in significantly less calcium flux than WT (P < .001) (Figure 1H). Notably, the PAR4 S67E and R68E mutants induced minimal calcium flux above baseline, such that we cannot exclude other minor CatG cleavage sites indicated by our mass spectrometry data (Figure 1H; supplemental Figure 5; and data not shown) could induce receptor activation, as has been shown with elastase-cleavage of PAR2.19 Since neighboring amino acids can be critical in protease cleavage,20 a Ser66 (S66E) mutant was generated that showed a slight decrease in CatG-induced calcium flux compared with WT and a significantly greater calcium flux than mock transfection (Figure 1H). None of the PAR4 mutants were significantly different than WT when stimulated by thrombin in the presence of a PAR1 inhibitor (Figure 1I). Taken together, these data support CatG, but not thrombin, cleavage of PAR4 between Ser67-Arg68.
Functional characterization of CatG-generated PAR4 tethered ligand
A synthetic peptide RALLLGWVPTR representing the CatG-generated PAR4 tethered ligand was synthesized to assess its potential functionality. RALLLGWVPTR induced calcium flux in HEK cells transfected with WT PAR4 compared with mock, indicating the tethered ligand induces signaling through PAR4 (Figure 2A). Platelets treated with RALLLGWVPTR demonstrated increased integrin activation, α granule release, and platelet aggregation (Figure 2B-E). In addition, RALLLGWVPTR induced a significant and sustained calcium flux in platelets similar to other peptide-tethered ligands21 (Figure 2F). Peak calcium flux was similar between CatG and RALLLGWVPTR, and consistent with previous literature, CatG-induced platelet calcium flux decreased over time13 (supplemental Figure 6).
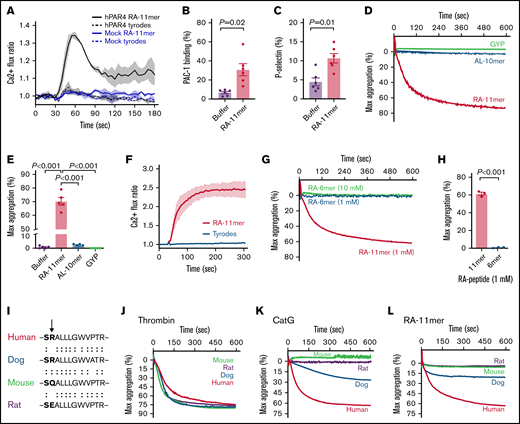
CatG-generated PAR4 tethered ligand RALLLGWVPTR induces platelet activation and aggregation. (A) WT human PAR4 (hPAR4, black) or empty vector (mock, blue) was expressed in HEK293T/17 cells and treated with 1.5 mM RALLLGWVPTR (RA-11mer, solid line) or tyrodes (dash lines). Thick lines and thin vertical lines are means and SEMs, respectively. n = 3 independent experiments performed in duplicate. (B-C) Washed platelets (n = 6) were treated with buffer or 1 mM RA-11mer, and platelet activation was measured by (B) PAC-1 binding (mean percent ± SEM) and (C) P-selectin expression (mean percent ± SEM). (D) Representative aggregation tracing of washed platelets treated with 1 mM GYPGQV (GYP), ALLLGWVPTR (AL-10mer), or RA-11mer. (E) Maximum (max) aggregation of platelets treated with buffer or 1 mM of each indicated peptide (mean percent ± SEM; n = 5). (F) Representative tracing of platelet calcium flux induced by RA-11mer (2 mM, black line). Tyrodes buffer (red) served as a negative control. n = 4 independent experiments performed in duplicate. (G) Representative tracing of washed platelets treated with 1 mM or 10 mM RA-6mer or 1 mM RA-11mer. (H) Quantification of maximum (max) aggregation (percent ± SEM) elicited by treatment of same subjects’ platelets with RA-11mer or RA-6mer (1 mM, n = 3). (I) Dog, human, mouse, and rat PAR4 sequence alignment of the 12 amino acids adjacent to the plasma membrane of the first (N-terminal) PAR4 extracellular domain. Arrow indicates Arg68 in humans where CatG cleaves PAR4. (J-L) Representative aggregation tracing of dog (blue), human (red), mouse (green), and rat (purple) washed platelets treated with 1 U/mL human thrombin (J), 1 µM human CatG (K), or 1 mM RA-11mer (L). n > 3 for human and mouse (J-L); n = 2 for dog and rat (J-K); n = 2 for rat (L); n = 1 for dog (L).
CatG-generated PAR4 tethered ligand RALLLGWVPTR induces platelet activation and aggregation. (A) WT human PAR4 (hPAR4, black) or empty vector (mock, blue) was expressed in HEK293T/17 cells and treated with 1.5 mM RALLLGWVPTR (RA-11mer, solid line) or tyrodes (dash lines). Thick lines and thin vertical lines are means and SEMs, respectively. n = 3 independent experiments performed in duplicate. (B-C) Washed platelets (n = 6) were treated with buffer or 1 mM RA-11mer, and platelet activation was measured by (B) PAC-1 binding (mean percent ± SEM) and (C) P-selectin expression (mean percent ± SEM). (D) Representative aggregation tracing of washed platelets treated with 1 mM GYPGQV (GYP), ALLLGWVPTR (AL-10mer), or RA-11mer. (E) Maximum (max) aggregation of platelets treated with buffer or 1 mM of each indicated peptide (mean percent ± SEM; n = 5). (F) Representative tracing of platelet calcium flux induced by RA-11mer (2 mM, black line). Tyrodes buffer (red) served as a negative control. n = 4 independent experiments performed in duplicate. (G) Representative tracing of washed platelets treated with 1 mM or 10 mM RA-6mer or 1 mM RA-11mer. (H) Quantification of maximum (max) aggregation (percent ± SEM) elicited by treatment of same subjects’ platelets with RA-11mer or RA-6mer (1 mM, n = 3). (I) Dog, human, mouse, and rat PAR4 sequence alignment of the 12 amino acids adjacent to the plasma membrane of the first (N-terminal) PAR4 extracellular domain. Arrow indicates Arg68 in humans where CatG cleaves PAR4. (J-L) Representative aggregation tracing of dog (blue), human (red), mouse (green), and rat (purple) washed platelets treated with 1 U/mL human thrombin (J), 1 µM human CatG (K), or 1 mM RA-11mer (L). n > 3 for human and mouse (J-L); n = 2 for dog and rat (J-K); n = 2 for rat (L); n = 1 for dog (L).
LC-MS/MS analysis also indicated a minor CatG cleavage between Arg68 and Ala69 under the conditions of our experiment, so we also synthesized an ALLLGWVPTR 10mer. However, platelets exposed to ALLLGWVPTR displayed no discernable platelet aggregation or calcium flux (Figure 2D-E; supplemental Figure 6) independent of peptide concentration (supplemental Figures 6 and 7). Notably, 1 mM RALLLGWVPTR produced maximal platelet aggregation, while 6 mM GYPGQV (the thrombin-generated PAR4 tethered ligand) was necessary to induce maximal platelet aggregation consistent with previous literature, demonstrating high concentrations of GYPGQV are needed to induce receptor activation (Figure 2D-E; supplemental Figure 7). The CatG-generated tethered ligand appears to be a more potent activator of PAR4 than the thrombin-generated tethered ligand, but the CatG enzyme is much less potent than the thrombin enzyme. Perhaps thrombin has greater affinity for the PAR4 extracellular domain than does CatG.
PAR4 signaling can be induced by the thrombin-tethered ligand with as few as 6 amino acids (GYPGQV), but a 6mer from the CatG tethered ligand (RALLLG) was not able to induce platelet aggregation (Figure 2G-H). We also synthesized an intermediate length peptide, RALLLGWV 8mer, but were unable to test its activating potential due to its inability to go into solution despite numerous attempts with different solvents. Thus, we cannot exclude the possibility that the minimal tethered ligand length required for PAR4 activation in vivo may be less than 11 residues. Having said that, perhaps the hydrophobic portion of the C-terminus of RALLLGWVPTR stabilizes the tethered ligand on the platelet plasma membrane.
Genetics of PAR4 variants
Conservation of amino acid sequence across species can be supportive and informative of sequence function. Supplemental Table 1 shows the sequence alignments of PAR4 N-terminal from 12 animals, 4 of which are shown in Figure 2I. Human thrombin induced platelet aggregation in humans, dogs, mice, and rats (Figure 2J), whereas human CatG only induced platelet aggregation in humans and dogs (Figure 2K). Similar to CatG, the RA-11mer induced aggregation only in humans and dogs (Figure 2L). Dog platelets were not as responsive to human CatG, perhaps due to inefficient binding of human CatG to dog PAR4 or the substitution of an alanine for a proline in the dog PAR4 sequence. Furthermore, the human RA-11mer could be less effective binding to the dog PAR4 extracellular domain that mediates the signaling response to the dog tethered ligand. This genetic data lends support to the mutagenesis and functional peptide data regarding the importance of Arg68 in mediating CatG-mediated PAR4 cleavage and activation. Perhaps the positive charge of Arg68 is important for the activity of the tethered ligand (mouse Gln is uncharged while rat Glu is negatively charged).
Implications
There are a relatively small number of serine protease-generated functional PAR tethered ligands. Identifying the potentially diverse array of PAR tethered ligands may be important if they activate different cellular signaling pathways or vary by pathophysiologic conditions. In certain clinical settings, such as those with extensive platelet-neutrophil interactions or leukocytosis in malignancy, CatG may dampen or eliminate thrombin signaling through PAR1 and PAR4, while the CatG tethered ligand could induce persistent PAR4 signaling.22 PAR4 is also expressed in nonplatelet tissues, where inflammation alters function and expression levels, and CatG could affect activation.23,24 Lastly, in hemorrhagic conditions, the RALLLGWVPTR peptide could be therapeutic for rescuing or enhancing platelet reactivity.
Acknowledgments
The authors wish to acknowledge Mark Cody for his technical assistance with experiments; Orvelin Roman, Ravi Ranjan, and Candace Reno for their assistance obtaining dog and rat platelets; Cynthia David from the University of Arizona, Analytical and Biological Mass Spectrometry Core Facility; James Cox from the University of Utah Mass Spectrometry Core for assistance with peptide analysis; and Guy Zimmerman for his thoughtful insight on experiments. The authors are grateful for figure preparation expertise from Diana Lim.
This work was supported by grants from the National Institutes of Health (R01HD093826 [C.C.Y.], R01HL102482 [P.F.B.], and K01AG059892 [R.A.C.]), the American Heart Foundation (21POST830138 [F.D.]), and an American Society of Hematology Bridge Grant (DSS-10060581 [P.F.B.]). Research reported in this publication was supported by the Flow Cytometry Core at the University of Utah and the National Center for Research Resources of the National Institutes of Health under Award Number 1S10RR026802-01.
Authorship
Contribution: M.L.S., P.F.B., and R.A.C. conceived and designed the experiments; M.L.S., I.B., F.D., J.W.R., J.A., K.P., M.T.N., C.C.Y., J.R.H., P.F.B., and R.A.C. performed experiments; M.L.S., I.B., K.P., P.F.B., and R.A.C. analyzed results and made the figures; M.L.S., P.F.B., and R.A.C. wrote the manuscript; and all authors reviewed and critically edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Robert A. Campbell, University of Utah Health Sciences Center Eccles Institute of Human Genetics, 15 North 2030 East, Building 533, Suite 4225, Salt Lake City, UT 84112; e-mail: rcampbell@u2m2.utah.edu.

