

Key Points
Src-related thrombocytopenia is an inherited syndrome that has immune thrombocytopenia-like features.
A constitutive upregulation of the B-cell receptor pathway may underlie the autoimmune manifestations of the disease.

Abstract
Src-related thrombocytopenia (SRC-RT) is a rare autosomal dominant, inherited platelet disorder resulting from the p.E527K heterozygous germline gain-of-function variant of Src. To date, genetic diagnosis of the disease has only been reported in 7 patients from 3 unrelated families. The clinical features ranged from isolated thrombocytopenia to complex syndromic manifestations characterized by thrombocytopenia, bleeding, myelofibrosis, splenomegaly, and bone disease. We report a new 3-generation kindred with the Src p.E527K variant. Patients presented with rather variable platelet counts (38-139 × 109/L), mildly impaired platelet function, >15% immature platelet fraction, and with a significant proportion of large-giant platelets. Four adults from the family were diagnosed with immune thrombocytopenia (ITP) and underwent splenectomy, achieving sustained platelet counts >75 × 109/L for several years; increases in platelet counts were also observed after corticosteroid therapy. Four of 7 Src p.E527K variant carriers showed immune defects and recurrent infections. In addition, a range of neurological symptoms, from specific language impairment to epilepsy, was seen in some family members. Patient platelets exhibited constitutive Src, Bruton tyrosine kinase, and phospholipase Cγ2 activation, and after stimulating CD19 cells by crosslinking surface immunoglobulin M, phosphorylated extracellular signal-regulated kinase (ERK) was significantly increased in B cells from individuals carrying the Src p.E527K substitution. In summary, in addition to causing impaired platelet production, SRC-RT may associate immune dysregulation and increased platelet consumption. In families in whom several members are responsive to ITP-directed therapies, an underlying Src p.E527K variant should be excluded.
Introduction
Src family kinases (SFKs) are nonreceptor protein-tyrosine kinases that regulate signal transduction. This involves modulation by SFKs of several signaling pathways involved in cell differentiation, proliferation, survival, motility, and adhesion. The human SFK consist of 8 typical family members (Src, Fyn, Yes, Fgr, Hck, Blk, Lck, and Lyn) and 3 atypical family members (Brk, Frk, Srms).1 Src, Fyn, and Yes are expressed ubiquitously, whereas the other SFKs show tissue-specific expression. Src is the most abundant SFK in human platelets, followed by Lyn and Fyn. They play a central role in mediating the rapid response of platelets to vascular injury. Src and Fyn are positive regulators of platelet activation, whereas Lyn plays a dual role.2 Beside platelets, Src is highly expressed in osteoclasts and neurons.3-5 In all SFKs, 2 essential phosphorylation sites are highly conserved, 1 in the activation loop and the second in the C-terminal tail. Phosphorylation of Tyr419 in the activation loop of the kinase domain upregulates enzyme activity, whereas phosphorylation of Tyr530 in the C-terminal tail decreases enzyme activity by blocking of the catalytic site necessary for kinase activation.6
Src is constitutively associated with αIIbβ3, is required for αIIbβ3-dependent activation of Syk, and is also part of the GPIb-IX-V signaling pathways.2 Leukocyte SFKs play an important role in the regulation of many activation responses, especially in inflammatory responses. SFKs are present in immune cells with dominant expression of Lck and Fyn in T cells and natural killer cells; Lyn, Fyn, and Blk in B cells and mast cells; and Hck, Fgr, and Lyn in myeloid cells such as neutrophils and macrophages.7 In normal human T cells8 and B cells,9 Src is detected only following activation. In B cells, activated Src is necessary for the phosphorylation of STAT5b, which ultimately leads to B-cell proliferation.10
Src is an upstream regulator of Bruton tyrosine kinase (BTK).11,12 Increasing evidence shows that overexpression of BTK induces a spontaneous autoimmune phenotype with increased cytokine production.13-17 Also, gain-of-function (GOF) variants in tyrosine kinases downstream of Src can cause immune dysregulation and systemic inflammation.18-20 This raises the question of whether increased activation of Src may also cause immune dysregulation.
In 2016, Turro et al first identified a germline GOF variant in Src, p.E527K, in a family presenting with thrombocytopenia. Affected patients showed juvenile myelofibrosis, splenomegaly, and bone diseases including mild facial dysmorphia and premature edentulism.21 Recently, the same variant was identified in 2 unrelated children with very different clinical features. One was a 5-year-old girl with a complex syndromic phenotype characterized by variable thrombocytopenia associated with facial dysmorphism, severe osteoporosis, and neurological disorders, such as autism and delayed language development.22 The second was a 2-year-old boy born to healthy parents, with isolated nonsyndromic thrombocytopenia.23
Here, we describe a previously unreported large pedigree with Src-related thrombocytopenia (SRC-RT). These patients present with autosomal dominant thrombocytopenia and autoimmune features, likely related to impaired megakaryocytopoiesis and disturbed B-cell receptor (BCR) signaling.
Patients
We studied an extended family with 7 members affected with the Src p.E527K heterozygous germline GOF variant, thrombocytopenia, and a high frequency of autoimmune and neurological disorders (Figure 1A; Table 1).
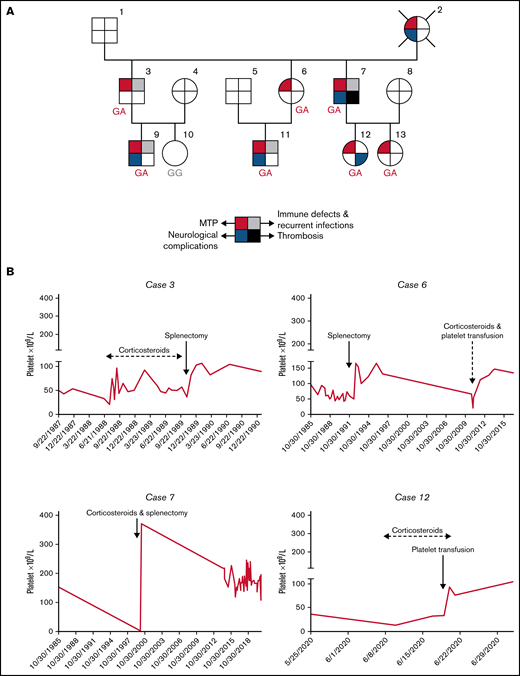
Pedigree of the family and platelet responses to therapeutic approaches. (A) Family pedigree and presence of SRC-RT resulting from Src p.E527K gain-of-function mutation. The upper left quarter burgundy shading in symbols indicates individuals with macrothrombocytopenia (MTP), the lower left quarter blue shading denotes neurological complications, the upper right quarter gray shading indicates immune defects (other than ITP) and recurrent infections, and the lower right quarter black shading indicates thrombosis. The deceased patient is indicated by a slash symbol. Patients carrying the heterozygous c.1579G>A variant in SRC are identified with GA genotype, and noncarriers with GG genotype. (B) Platelet responses to splenectomy and/or corticosteroids. The graphs show the timing of splenectomy and corticosteroid therapy and platelet count profiles of patients 3, 6, 7, and 12.
Pedigree of the family and platelet responses to therapeutic approaches. (A) Family pedigree and presence of SRC-RT resulting from Src p.E527K gain-of-function mutation. The upper left quarter burgundy shading in symbols indicates individuals with macrothrombocytopenia (MTP), the lower left quarter blue shading denotes neurological complications, the upper right quarter gray shading indicates immune defects (other than ITP) and recurrent infections, and the lower right quarter black shading indicates thrombosis. The deceased patient is indicated by a slash symbol. Patients carrying the heterozygous c.1579G>A variant in SRC are identified with GA genotype, and noncarriers with GG genotype. (B) Platelet responses to splenectomy and/or corticosteroids. The graphs show the timing of splenectomy and corticosteroid therapy and platelet count profiles of patients 3, 6, 7, and 12.
Biological and clinical features of Src p.E527 carriers
| ID | Sex | Age at diagnosis | Bleeding score | Platelet count at diagnosis (×109/L) | Last platelet count (×109/L) (% IPF) | Splenomegaly/splenectomy | Neurological complications | Immune defects | Relevant infection complications | Thrombosis |
|---|---|---|---|---|---|---|---|---|---|---|
| 2* | F | Unknown | Unknown | Unknown | 203 (Nr) | Yes/yes (<30 y) | Epilepsy; SUDEP | Unknown | Unknown | Unknown |
| 3 | M | 3 y | 9 | 57 | 83 (Nr) | Yes/yes (13 y) | No | Plaque psoriasis; uvular angioedema; partial IgM deficiency | Recurrent tonsillitis >2 episodes/y | No |
| 6 | F | 8 y | 1 | 96 | 134 (27.2) | Yes/yes (14 y) | No | No | Pityriasis versicolor | No |
| 7 | M | 6 y | 4 | 98 | 177 (17.6) | Yes/yes (21 y) | Complex epileptic seizures | Psoriasis; severe bronchial asthma; ileal Crohn disease; autoimmune pleuropericarditis; partial IgM deficiency | Upper respiratory (>1/y); epididymo-orchitis. Pityriasis versicolor; papulopustular acne | Portal thrombosis and pulmonary embolism; jugular vein thrombosis |
| 9 | M | 6 d | 3 | 38 | 17 (36.9) | Yes/no | Megacisterna magna (asymptomatic) | Intrinsic bronchial asthma; partial IgA deficiency | Recurrent bronchitis (>3/y). Pityriasis versicolor | No |
| 11 | M | 3 d | 3 | 70 | 66 (16.1) | NR/no | Speech and language impairment | Atopic dermatitis; bronchial hyperreactivity | Recurrent bronchitis, >2 episodes/y | No |
| 12 | F | 6 y | 4 | 71 | 105 (40.0) | NR/no | Anxiety and behavior abnormalities | No | Otitis media; upper respiratory (<2 episodes/y) | Deep vein thrombosis |
| 13 | F | 10 y | 0 | 139 | 114 (NR) | NR/no | No | No | Upper respiratory (>2 episodes/y) | No |
| ID | Sex | Age at diagnosis | Bleeding score | Platelet count at diagnosis (×109/L) | Last platelet count (×109/L) (% IPF) | Splenomegaly/splenectomy | Neurological complications | Immune defects | Relevant infection complications | Thrombosis |
|---|---|---|---|---|---|---|---|---|---|---|
| 2* | F | Unknown | Unknown | Unknown | 203 (Nr) | Yes/yes (<30 y) | Epilepsy; SUDEP | Unknown | Unknown | Unknown |
| 3 | M | 3 y | 9 | 57 | 83 (Nr) | Yes/yes (13 y) | No | Plaque psoriasis; uvular angioedema; partial IgM deficiency | Recurrent tonsillitis >2 episodes/y | No |
| 6 | F | 8 y | 1 | 96 | 134 (27.2) | Yes/yes (14 y) | No | No | Pityriasis versicolor | No |
| 7 | M | 6 y | 4 | 98 | 177 (17.6) | Yes/yes (21 y) | Complex epileptic seizures | Psoriasis; severe bronchial asthma; ileal Crohn disease; autoimmune pleuropericarditis; partial IgM deficiency | Upper respiratory (>1/y); epididymo-orchitis. Pityriasis versicolor; papulopustular acne | Portal thrombosis and pulmonary embolism; jugular vein thrombosis |
| 9 | M | 6 d | 3 | 38 | 17 (36.9) | Yes/no | Megacisterna magna (asymptomatic) | Intrinsic bronchial asthma; partial IgA deficiency | Recurrent bronchitis (>3/y). Pityriasis versicolor | No |
| 11 | M | 3 d | 3 | 70 | 66 (16.1) | NR/no | Speech and language impairment | Atopic dermatitis; bronchial hyperreactivity | Recurrent bronchitis, >2 episodes/y | No |
| 12 | F | 6 y | 4 | 71 | 105 (40.0) | NR/no | Anxiety and behavior abnormalities | No | Otitis media; upper respiratory (<2 episodes/y) | Deep vein thrombosis |
| 13 | F | 10 y | 0 | 139 | 114 (NR) | NR/no | No | No | Upper respiratory (>2 episodes/y) | No |
ID, number in family pedigree (Figure 1A); F, female; IPF, immature platelet fraction; M, male; NR, not recorded; SUDEP, sudden unexpected death in epilepsy.
Died.
Patient 2
A female born in 1955 was diagnosed with immune thrombocytopenia (ITP) and splenectomized with complete response (platelet count: 203 × 109/L). She was also diagnosed with idiopathic generalized epilepsy and died at age 42 years from sudden unexpected death in epilepsy.
Patient 3
A male born in 1976 was diagnosed at the age of 9 years with ITP, with a platelet count of 57 × 109/L. Three years after diagnosis, the platelet count was 22 × 109/L, and treatment with 34 mg/d of methylprednisolone was started. After 2 weeks, platelet counts increased to 73 × 109/L, and from July 1988 to November 1989 he received continuous steroid treatment (range, 4-32 mg/d) with a dose-dependent platelet count response (Figure 1B; supplemental Figure 1). In November 1989, he underwent splenectomy, with histological findings of congestive large spleen (14 × 9 × 5.5 cm). Since then, his platelet counts are above 75 × 109/L without additional treatment. Indicators of immune dysfunction are a partial selective immunoglobulin M (IgM) deficiency (31 mg/dL; normal range [NR], 40-275 mg/dL), and mild hypogammaglobulinemia (7 g/L; NR, 8-14 g/L). During follow-up, he developed recurrent tonsillitis, plaque psoriasis, and uvular angioedema (Table 1).
Patient 6
A female born in 1977 was diagnosed with ITP at the age of 8 years (platelet count at diagnosis: 96 × 109/L). Over the following 7 years, her platelet counts slowly declined. She underwent splenectomy in 1992 (platelet count at surgery: 58 × 109/L) with histological findings of congestive spleen with occasional megakaryocytes noted. Thereafter, her platelet counts remained >100 × 109/L until December 2010 (week 36 of her pregnancy) when platelet counts declined to 22 × 109/L. At that time, 60 mg/d of prednisone was started, prompting an increase in platelet counts (48 × 109/L) within 1 week. She was transfused with 1 platelet concentrate and had an uneventful delivery. Since then, the patient maintained platelets above 100 × 109/L, with 27.3% immature platelet fraction (IPF) (IPF NR <7%) (Figure 1B). No other autoimmune features have been recorded.
Patient 7
A male born in 1979 was diagnosed with ITP at the age of 6 years (platelet count at diagnosis, 98 × 109/L). In January 2020, he was started on 48 mg/d of methylprednisolone, at a platelet count of 4 × 109/L. After 9 weeks, the platelet count increased to 91 × 109/L, and the patient underwent splenectomy (spleen of 18 × 12 × 8 cm). Since then, platelet counts have remained above 150 × 109/L (Figure 1B) with IPF between 13.1% and 22.6%. In addition to mild IgM and gammaglobulin deficiency (35 mg/dL and 8 g/L, respectively), he presented with symptoms of immune dysregulation such as psoriasis, severe persistent bronchial asthma, ileal Crohn disease, and autoimmune pleuro-pericarditis (Table 1). The patient has also suffered from various vascular events such as portal thrombosis, pulmonary embolism, and jugular vein thrombosis. Since 2000, he has presented complex, partial epileptic seizures with a frequency of 6 to 8 per month, and in January 2018 he underwent right anterior and medial temporal lobe resection with residual cognitive impairment and reduced but persistent (2-3 per month) seizure episodes.
Patient 9
A male born in 2001 was diagnosed with inherited thrombocytopenia, with a platelet count of 38 × 109/L, and with mega-cisterna magna at the age of 6 days, without subsequent associated neurological impairment. At present, the patient maintains platelet counts below 40 × 109/L with an IPF of 30.3% to 35.6%. In the absence of hemorrhagic symptoms, he has not received specific treatment for thrombocytopenia. Ultrasound evaluation identified a mildly enlarged spleen (14.3 cm) at age 19 years. Biochemical and hematological features indicate partial IgA deficiency (41 mg/dL; NR, 70-400 mg/dL), mild hypogammaglobulinemia (6 g/L), and a reduction in the CD19+ CD27+ IgM-IgD-B memory switched plasmablast subpopulation (5.21%; NR, 6.5%-29.1%). He suffers recurrent asthma and bronchitis episodes.
Patient 11
A male born in 2010 was diagnosed with inherited thrombocytopenia at the age of 3 days, with a platelet count of 53 × 109/L and IPF 16%. Subsequently, a mild, unspecified developmental disorder of speech and language was found. At the age of 10 years, he required hospital admission for severe bronchospasm in the context of influenza A infection. He showed epistaxis, and a course of 0.3 mg/kg dexamethasone was administered. Platelet count at admission was 16 × 109/L and rose to 38 × 109/L and to 66 × 109/L after 4 and 10 days of steroid therapy, respectively. Since then, he has not required any additional treatment for thrombocytopenia.
Patient 12
A female born in 1999 was diagnosed with inherited thrombocytopenia at the age of 6 years, with a platelet count of 71 × 109/L. At the age of 17 years, she presented with spontaneous lower limb vein thrombosis and received anticoagulation with acenocoumarol for 10 months. Thrombophilia studies (antithrombin, protein C, protein S, factor V Leiden, prothrombin G20210A, lupus anticoagulant) were normal. Her platelet counts have remained ∼80 × 109/L (IPF, 24%-40%). In June 2020, during the eighth month of her pregnancy, platelet levels dropped to 14 × 109/L. She received 1 mg/kg per d prednisone. Within 1 week, platelet counts increased to 34 × 109/L; she received a platelet transfusion and underwent labor with a platelet count of 93 × 109/L (Figure 1B). The patient did not require other platelet increasing drugs and is currently receiving medication for anxiety and behavior abnormalities only.
Patient 13
A female born in 2009 was diagnosed as being a carrier of the Src p.E527K heterozygous variant. Her medical history does not reveal other relevant diseases. Platelet count at diagnosis and during follow-up were only mildly reduced (100-140 × 109/L).
Patient 10
This patient, who was used as a control, was confirmed not to be a carrier of the Src p.E527K variant and to have neither thrombocytopenia nor immune dysfunction.
Materials and methods
The members of this family were recruited through the Spanish multicenter project “Functional and Molecular Characterization of Patients with Inherited Platelet Disorders.” This project obtained approval from the Ethics Committee of the Hospital Reina Sofia (Murcia, Spain) and follows the Declaration of Helsinki. All participants gave written informed consent. Information on the deceased patient was obtained following verbal consent from her relatives. Clinical records were reviewed, and the bleeding symptoms were scored using the International Society on Thrombosis and Haemostasis bleeding assessment tool (ISTH-BAT).24
Blood samples were drawn into EDTA and sodium citrate tubes; serum was obtained from nonanticoagulated blood. DNA from 2 index patients (3 and 6) was analyzed by a predesigned high-throughput sequencing (HTS) gene panel25,26 using an Ion Torrent PGM platform (Thermo Fisher Scientific, Waltham, MA). Sequences were annotated and candidate genetic variants were identified as reported elsewhere.25,26 Selected variants were confirmed in these patients, and segregation in the family was confirmed by means of DNA polymerase chain reaction amplification with specific primers and Sanger sequencing using an ABI 3130 automated sequencer. In selected family members, the BTK gene was analyzed using nanopore sequencing technology (supplemental Methods).
The immature platelet fraction (IPF) was quantified using Sysmex XE-2100 (Sysmex, Sant Just Desvern, Spain). Plasma levels of various cytokines were measured using Luminex xMAP technology with a customized MILLIPLEX kit (HCYTA-60K, Millipore, Madrid, Spain) that included 14 cytokines (interleukin-1α [IL-1α], IL-1β, IL-1ra, IL-2, IL-6, IL-8, IL-10, IL-12 [p40], IL-12 [p70], IL-15, IL-18, monocyte chemoattractant protein-1/CCL2, macrophage-colony-stimulating factor, and tumor necrosis factor-α). The analysis was performed in a MAGPIX system (Luminex; Thermo Fisher, San Cugat del Valles, Spain) using the xPONENT software, and the concentrations were obtained using the Milliplex Analyst v.5.2 Flex software (VigeneTech, Rockville, MD). Sera of patients were tested with an enzyme‐linked immunosorbent assay for antiplatelet antibodies (PAKPLUS, Immucor, Cerdanyola del Valls, Spain).
Platelet surface glycoprotein (GP) expression, agonist-induced fibrinogen binding, and α- and δ-granule secretion (CD62 and CD63, respectively) were assessed by flow cytometry.26,27 Light transmission aggregometry and platelet examination by transmission electron microscopy were carried out as described.26-29 Immunofluorescence staining of α-granule proteins (thrombospondin, von Willebrand factor [VWF], and P-selectin), δ-granule markers (LAMP-1, LAM-2, and CD63), β1-tubulin, Src, and phosphorylated-Src were performed on blood smears as reported30 using specific antibodies (anti-thrombospondin antibody [ab85762, Abcam, Cambridge, UK], anti-P-Selectin [555522, BD Biosciences, San Jose, CA], anti-VWF [A0082, Dako, Waldbronn, Germany], anti-LAMP1 [sc18821, Santa Cruz Biotechnology, Heidelberg, Germany], anti-LAMP2 [sc18822, Santa Cruz Biotechnology], anti-CD63 [558019, BD Biosciences], anti-β1-tubulin [T4026; Merck Life Science, Darmstadt, Germany], anti c-Src [17AT28, sc130124; Santa Cruz Biotechnology], and anti-Tyr-419-phosphorylated-c-Src [sc-139601, Santa Cruz Biotechnology]). In resting platelets, protein content and tyrosine phosphorylation of specific targets were evaluated in platelet lysates by standard immunoblotting26 using Src (Santa Cruz Biotechnology), Phospho-Src (Santa Cruz Biotechnology), BTK, Phospho-BTK (Tyr223), Y759 phospholipase Cγ2 (PLCγ2) antibodies (Cell Signaling Technology; Frankfurt, Germany), and PLCγ2 antibody (Santa Cruz Biotechnology).
Human peripheral blood mononuclear cells were isolated from fresh blood samples of cases 3 and 9, and healthy controls by Ficoll-Paque (GE Healthcare Life Sciences) density centrifugation method. Peripheral blood mononuclear cells were stained with anti-CD19 and stimulated with 20 μg/mL soluble anti-IgM antibody (Jackson Immunoresearch Labs, West Grove, PA) at 37°C for 5 minutes. Cells were fixed and permeabilized using the Intrasure kit (BD), and incubated with P-Syk, P-BTK, and P-ERK (Cell Signaling Technology) antibodies. Following a washing step, a goat anti-rabbit IgG (H+L chains) secondary antibody Alexa Fluor 633 was added (Invitrogen), and the analysis was performed using a BD ACCURI cytometer.
Results
Platelet counts and morphology, and bleeding score
The 7 available patients in the pedigree (Figure 1A; Table 1) with lifelong thrombocytopenia had platelet counts at diagnosis that ranged from 38 to 139 × 109/L (NR, 142-359 × 109/L [n = 107]). The IPF was consistently increased (Table 1), and the mean platelet volume provided by the hematological counter was normal or slightly increased, but examination of peripheral blood smears showed platelet anisocytosis (supplemental Figure 2). No other abnormalities were observed in hematological parameters, except for mild iron deficiency anemia in 2 patients (7 and 12). The median ISTH-BAT score in the patients was 3 (range, 1-4) (Table 1).
Molecular studies
DNA analysis with a HTS-gene panel in cases 3 and 6 identified 3 candidate variants in genes known to be responsible for inherited thrombocytopenia: the heterozygous c.1579G>A variant (p.E527K) in the SRC gene (OMIM 616937, LRG_1018, NM_198291.2); the heterozygous c.754A>G variant (p.I252V) in the ITGB3 gene (OMIM 273800, LRG_481, NM_000212.2); and the heterozygous c.236C>T variant (p.T79I) in the GP9 gene (OMIM 231200; LRG_477, NM_000174.4). Sanger sequencing confirmed the presence of these variants in the 2 probands, but only the SRC variant was found to segregate with thrombocytopenia in 5 additional members of the family (patients 7, 9, 11, 12, and 13; Figure 1A). This variant was absent in the unaffected case 10 (Figure 1A). Nanopore sequencing of the BTK gene (15 of 22 kb) was analyzed in Src p.E527K variant carriers (cases 3, 6, 7, 9, 11, 12, and 13 in Figure 1A). No pathogenic variants were detected.
Platelet studies
As illustrated in Figure 2A and supplemental Table 1, electron microscopy analysis showed round and significantly enlarged platelets with slightly but not significantly reduced numbers of granules in individuals with the Src p.E527K variant. Aggregometry assays were performed in 3 affected individuals. As compared with a healthy control, patients displayed an impairment of platelet aggregation in response to arachidonic acid and collagen related peptide, and to a lesser extent to adenosine diphosphate and thrombin receptor activating peptide (Figure 2B). Flow cytometry analysis showed normal expression of GP Ib/IX and Ia, slightly increased GPIIb/IIIa (αΙΙbβ3 integrin), and reduced GPVI levels in Src p.E527K carriers (Figure 3). Agonist-induced fibrinogen binding, and α- and δ-granule secretions were markedly impaired in Src p.E527K platelets (Figure 3).
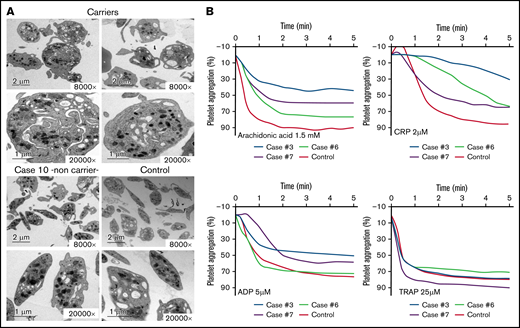
Electron microscopy and aggregation studies in carriers and noncarriers of the Src p.E527K variant. (A) Electron microscopy of platelets from 2 carriers of the Src p.E527K variant (patients 3 and 7), a noncarrier family member (patient 10), and a healthy control. Magnification for each image is shown (8000-20 000×). Images were acquired in a JEOL transmission electron microscope. (B) Platelet-rich plasma aggregation profiles in response to the indicated agonists from patients 3, 6, 7, and a control subject. CRP, collagen-related peptide; TRAP, thrombin receptor activating peptide (PAR-1).
Electron microscopy and aggregation studies in carriers and noncarriers of the Src p.E527K variant. (A) Electron microscopy of platelets from 2 carriers of the Src p.E527K variant (patients 3 and 7), a noncarrier family member (patient 10), and a healthy control. Magnification for each image is shown (8000-20 000×). Images were acquired in a JEOL transmission electron microscope. (B) Platelet-rich plasma aggregation profiles in response to the indicated agonists from patients 3, 6, 7, and a control subject. CRP, collagen-related peptide; TRAP, thrombin receptor activating peptide (PAR-1).
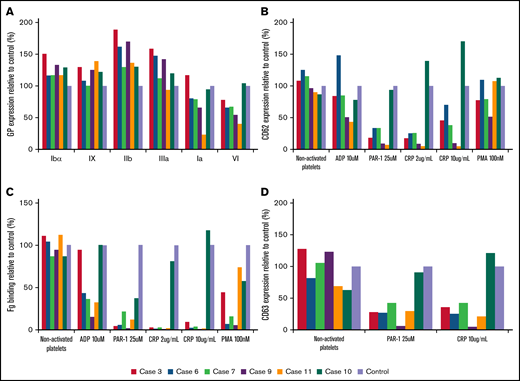
Flow cytometric analysis of platelet glycoproteins, fibrinogen binding, and granule secretion. (A) Platelet glycoprotein expression of Src p.E527K carriers (cases 3, 6, 7, 9, and 11) and a control. (B) Fibrinogen-Alexa488 binding, (C) α granule secretion, and (D) δ granule secretion in resting state and upon platelet stimulation with specific agonists. The graphs show median fluorescence intensity relative to the control (100%). Fg, fibrinogen.
Flow cytometric analysis of platelet glycoproteins, fibrinogen binding, and granule secretion. (A) Platelet glycoprotein expression of Src p.E527K carriers (cases 3, 6, 7, 9, and 11) and a control. (B) Fibrinogen-Alexa488 binding, (C) α granule secretion, and (D) δ granule secretion in resting state and upon platelet stimulation with specific agonists. The graphs show median fluorescence intensity relative to the control (100%). Fg, fibrinogen.
Platelet granule markers, as well as other proteins, were also evaluated in conventional blood smears by immunofluorescence with specific antibodies. Overall, Src p.E527K platelets showed mild-moderate impairment in the staining of the α-granule markers thrombospondin, VWF, and P-selectin (supplemental Figure 3). In contrast, the δ-granule markers, LAMP-1, LAMP-2, and CD63, were similarly expressed to control platelets (supplemental Figure 4). Interestingly, Src p.E527K platelets, especially those that were enlarged, also showed disturbed β1-tubulin staining (supplemental Figure 4). Because the Src p.E527K variant has been previously reported as a GOF mutation causing constitutive Src activation,21-23 we evaluated by western blot Src p.E527K platelets for the basal level of whole and phosphorylated Src at Tyrosine 419. The phospho-Y519 Src antibody may exhibit some level of off-target reactivity with other Src family members (Lyn, Fyn, Lck, Yes, and Hck) when phosphorylated at equivalent sites. However, antibodies to the phospho-Y519 Src or to total Src stained a band with the same molecular weight in platelet lysates. This suggests that patient platelets displayed increased Src phosphorylation, whereas total levels of the protein were similar to those in controls (Figure 4A). Because phospho-Src phosphorylates tyrosine residues in BTK, and phospho-BTK causes activation of PLCγ2, we tested whether the signaling pathways downstream of Src were tyrosine phosphorylated in resting platelets. As shown in Figure 4B, in the absence of external stimuli, Y223 phosphorylation of BTK was upregulated in Src p.E527K carrier platelets, but total levels of the protein were similar to that of control cells. Similar results were obtained for Y1217 PLCγ2, whereas no differences were observed in the levels of total protein content (Figure 4C).
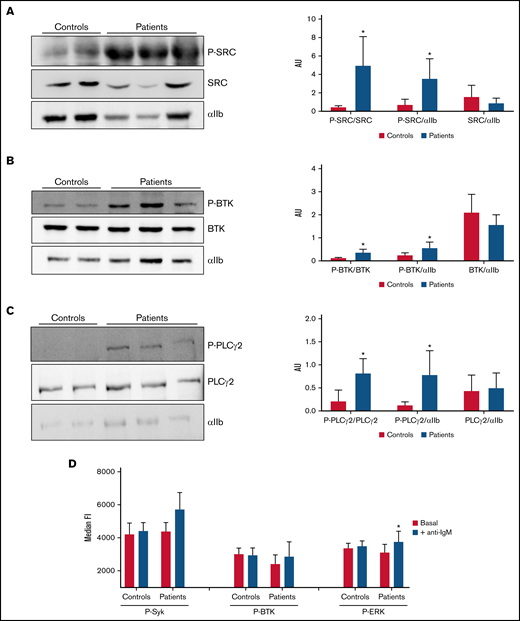
Analysis of tyrosine-phosphorylated proteins in platelets and lymphocytes. (A-C) Evaluation of resting platelet lysates was done in members of the SRC-RT pedigree carrying the Src p.E527K variant (patients 3, 6, and 7), in parallel with those of 2 controls. αIIb was used as loading control. *P ≤ .05 between patients and controls. (A) Representative image of immunoblotting experiments of SRC and phospho-SRC (P-SRC), and densitometric analysis of the bands obtained with 2 separate experiments (mean ± standard deviation). (B) Representative image of immunoblotting experiments of BTK and phospho-BTK (P-BTK), and densitometric analysis of the bands obtained with 2 separate experiments (mean ± standard deviation). (C) Representative image of immunoblotting experiments of PLCγ2 and phospho-PLCγ2 (P- PLCγ2), and densitometric analysis of the bands obtained with 2 separate experiments (mean ± standard deviation). (D) Flow cytometric evaluation of SRC downstream targets in B (CD19+) lymphocytes. Phosphorylation of tyrosine kinases were analyzed in unstimulated cells (white bars) or after stimulation with anti-IgM (black bars) in Src p.E527K carriers (patients 3 and 9) and control lymphocytes (n = 2) by flow cytometry. Data are representative of 2 individual experiments. Results reflect mean ± standard deviation values. AU, arbitrary units; FI, fluorescence intensity. *P ≤ .05 between stimulated and unstimulated samples.
Analysis of tyrosine-phosphorylated proteins in platelets and lymphocytes. (A-C) Evaluation of resting platelet lysates was done in members of the SRC-RT pedigree carrying the Src p.E527K variant (patients 3, 6, and 7), in parallel with those of 2 controls. αIIb was used as loading control. *P ≤ .05 between patients and controls. (A) Representative image of immunoblotting experiments of SRC and phospho-SRC (P-SRC), and densitometric analysis of the bands obtained with 2 separate experiments (mean ± standard deviation). (B) Representative image of immunoblotting experiments of BTK and phospho-BTK (P-BTK), and densitometric analysis of the bands obtained with 2 separate experiments (mean ± standard deviation). (C) Representative image of immunoblotting experiments of PLCγ2 and phospho-PLCγ2 (P- PLCγ2), and densitometric analysis of the bands obtained with 2 separate experiments (mean ± standard deviation). (D) Flow cytometric evaluation of SRC downstream targets in B (CD19+) lymphocytes. Phosphorylation of tyrosine kinases were analyzed in unstimulated cells (white bars) or after stimulation with anti-IgM (black bars) in Src p.E527K carriers (patients 3 and 9) and control lymphocytes (n = 2) by flow cytometry. Data are representative of 2 individual experiments. Results reflect mean ± standard deviation values. AU, arbitrary units; FI, fluorescence intensity. *P ≤ .05 between stimulated and unstimulated samples.
Antiplatelet antibodies and cytokine profile
In 1 (patient 7) of 5 Src p.E527K carriers (patients 3, 6, 7, 9, and 11), we found weak but broad reacting antiplatelet antibodies (GPIIb/IIIa, Ia/IIa, Ib/IX, IV, and HLA; OD ratio [OD value/cutoff] < 3. In addition, we also found anti-GPIIb/IIIa antibodies only in 1 of 3 sera from patients with ITP (included as positive controls).
Considering the inflammatory and immunodeficiency features of Src p.E527K carriers, we tested their inflammatory cytokine milieu (patients 3, 6, 7, 9, and 11). We found a significant decrease in plasma inflammatory biomarkers (IL-1a, IL-2, and tumor necrosis factor-α), compared with healthy individuals. In 5 individuals with newly diagnosed ITP, we confirmed significantly increased plasma levels of pro- (IL-6, monocyte chemoattractant protein-1, and macrophage-colony-stimulating factor) and anti- (IL-10) inflammatory cytokines, compared with healthy controls (Table 2).
Inflammatory cytokine profile of patients with SRC-RT
| pg/mL | Controls (n = 9) | Patients with ITP (n = 5) | Patients with SRC-RT (n = 5) |
|---|---|---|---|
| IL-1a | 6.03 ± 4.91 | 11.86 ± 6.33 | 2.68 ± 0.00*,† |
| IL-1b | 7.06 ± 7.07 | 7.62 ± 5.77 | 0.99 ± 0.53† |
| IL-1RA | 2.72 ± 2.44 | 3.82 ± 1.45 | 1.79 ± 1.97 |
| IL-2 | 0.46 ± 0.24 | 0.33 ± 1.39 | 0.30 ± 0.00* |
| IL-6 | 1.63 ± 1.83 | 10.94 ± 2.40‡ | 0.83 ± 1.07† |
| IL-8 | 0.76 ± 0.43 | 2.28 ± 1.40 | 0.75 ± 0.38 |
| IL-10 | 0.62 ± 0.00 | 2.92 ± 5.23‡ | 0.62 ± 0.00† |
| IL-12 p40 | 28.27 ± 12.53 | 17.03 ± 8.31 | 40.18 ± 35.87 |
| IL-12 p70 | 5.31 ± 4.21 | 2.49 ± 13.40 | 0.73 ± 0.00† |
| IL-15 | 5.63 ± 2.75 | 9.63 ± 3.35 | 4.01 ± 2.04† |
| IL-18 | 22.05 ± 10.32 | 23.75 ± 31.09 | 28.95 ± 12.39 |
| MCP-1 | 133.42 ± 46.72 | 200.14 ± 59.40‡ | 119.00 ± 13.14† |
| M-CSF | 46.45 ± 42.76 | 137.87 ± 152.15‡ | 51.27 ± 27.98† |
| TNF-α | 48.85 ± 36.95 | 49.65 ± 12.83 | 15.57 ± 6.79*,† |
| pg/mL | Controls (n = 9) | Patients with ITP (n = 5) | Patients with SRC-RT (n = 5) |
|---|---|---|---|
| IL-1a | 6.03 ± 4.91 | 11.86 ± 6.33 | 2.68 ± 0.00*,† |
| IL-1b | 7.06 ± 7.07 | 7.62 ± 5.77 | 0.99 ± 0.53† |
| IL-1RA | 2.72 ± 2.44 | 3.82 ± 1.45 | 1.79 ± 1.97 |
| IL-2 | 0.46 ± 0.24 | 0.33 ± 1.39 | 0.30 ± 0.00* |
| IL-6 | 1.63 ± 1.83 | 10.94 ± 2.40‡ | 0.83 ± 1.07† |
| IL-8 | 0.76 ± 0.43 | 2.28 ± 1.40 | 0.75 ± 0.38 |
| IL-10 | 0.62 ± 0.00 | 2.92 ± 5.23‡ | 0.62 ± 0.00† |
| IL-12 p40 | 28.27 ± 12.53 | 17.03 ± 8.31 | 40.18 ± 35.87 |
| IL-12 p70 | 5.31 ± 4.21 | 2.49 ± 13.40 | 0.73 ± 0.00† |
| IL-15 | 5.63 ± 2.75 | 9.63 ± 3.35 | 4.01 ± 2.04† |
| IL-18 | 22.05 ± 10.32 | 23.75 ± 31.09 | 28.95 ± 12.39 |
| MCP-1 | 133.42 ± 46.72 | 200.14 ± 59.40‡ | 119.00 ± 13.14† |
| M-CSF | 46.45 ± 42.76 | 137.87 ± 152.15‡ | 51.27 ± 27.98† |
| TNF-α | 48.85 ± 36.95 | 49.65 ± 12.83 | 15.57 ± 6.79*,† |
Inflammatory cytokines were assessed in plasma from patients with SRC-RT (n = 5; patients 3, 6, 7, 9, and 11), patients with ITP (n = 5), and healthy controls (n = 9) by using Luminex technology. All results express mean ± standard deviation.
MCP-1, monocyte chemoattractant protein-1; TNF-α, tumor necrosis factor-α.
P ≤ .05 for patients with SRC-RT vs controls.
P ≤ .05 for patients with SRC-RT vs those with ITP.
P ≤ .05 for patients with ITP vs controls.
Src downstream protein phosphorylation in lymphocytes
Upon BCR stimulation of human B cells, a signaling cascade is initiated whereby Src, Syk, BTK, and ERK are phosphorylated. As shown in Figure 4D, stimulation of B cells by crosslinking surface IgM resulted in higher levels of phosphorylated ERK in B cells from individuals carrying the Src p.E527K substitution (but not in controls) (19.95% vs 2.50%, respectively; P < .05). The basal phosphorylation level of Syk, BTK, and ERK did not differ between patients and controls.
Discussion
Our data confirm that dysregulation of Src signaling associates with impaired platelet function. Beside reduced collagen-induced aggregation, already reported in 3 unrelated pedigrees with p.E527K,21-23 these patients also showed an impaired aggregation response upon stimulation with agonists such as adenosine diphosphate and arachidonic acid. Similar to mice with ablated C-terminal Src kinase and increased platelet SFK activity,31 these patients present with macrothrombocytopenia and an increase in the proportion of reticulated, immature platelets. These data together with the reduced numbers of α-granules and alteration of the marginal band are likely explained by increased platelet turnover resulting from constitutively increased Src activity, in vivo platelet activation, and secondary exhaustion of platelets. In line with this assumption, in this family, despite thrombocytopenia, no relevant recurrent bleeding events were reported, and the ISTH-BAT scores were not consistently above normal ranges.32 On the contrary, several patients suffered from thrombotic events. Additionally, it has also been reported that Src p.E527K megakaryocytes exhibit perturbed interactions with the bone marrow matrix components.23 Thrombocytopenia associated with increased Src activation may therefore result from reduced proplatelet formation and increased platelet turnover.
In addition, data suggest that dysregulated immune mechanisms may also contribute to reduced platelet counts in patients with SRC-RT. The strongest evidence is the clinical observation that exposure to corticosteroids was consistently associated with an increase in platelet counts (patients 3, 6, 7, and 12). However, we could not present direct proof of an immune mechanism contributing to thrombocytopenia in this family. Although in the 4 patients in which splenectomy was performed, platelet counts increased, this might be also caused by reduced removal of activated platelets by the spleen. Similarly, the consistent IPF elevation seen in our patients (typically >15%, suggesting increased platelet turnover) has been observed both in ITP and in inherited macrothrombocytopenia.33 We found antiplatelet antibodies in the plasma only in 1 Src p.E527K carrier. However, this method is rather insensitive and neither confirms nor rules out ITP. Interestingly, patients carrying the Src p.E527K variant presented with a decrease in cytokines, whereas we found these cytokines elevated in newly diagnosed patients with ITP, in agreement with previous studies.34
Nevertheless, the presence of laboratory markers and/or clinical symptoms of immune dysregulation is striking in this family. We therefore addressed the phenotype of their immune cells. The crucial role of cytoplasmic tyrosine kinases in the complex signaling responsible for the activation of both innate and adaptive immune cells has been studied extensively.35,36 Overall, receptor crosslinking stimulates the enzymatic activation of receptor-bound SFK; these kinases play a cooperative role with SYK, its downstream kinase, whereby SYK communicates with BTK to phosphorylate and thus activate PLCγ2, which triggers the activation of ERK.37 Here, we show that upon IgM crosslinking, patients’ B cells showed a 19.9% increased ERK phosphorylation. In B lymphocytes, Lyn, but not Src, is the major SFK regulating the signaling events downstream the BCR.38 But B cells transduced with a constitutive active form of c-Src can increase phospho-STAT5b by 20%, which then mediates proliferation of these lymphocytes.10 It is therefore likely that the GOF mutation Src p.E527K also increases BCR signaling. In mice models, enhanced BCR signaling causes breakdown in self-tolerance and autoimmunity, preferentially in male animals.39 In humans, constitutive, BCR-autonomous signaling has been implicated in the pathogenesis of autoimmune diseases.40 Moreover, patients with downstream GOF mutations in SYK or PLCγ2, which are important for the BCR signaling pathway, show paradoxical features of immunodeficiency and hyperinflammatory responses.18-20 Similar to patients of this family, they have reduced serum levels of IgM, IgG, or IgA; decreased numbers of class-switched memory B cells; varying degrees of intestinal, skin, joint, and nervous system inflammation; and recurrent sinopulmonary infections.18-20 Moreover, patients with GOF mutations that lead to PLCγ2 activation present with the same increase in ERK phosphorylation upon anti-IgM stimulation of B cells41 as our patients, and their levels of IL-1 are substantially decreased compared with controls.19 In addition, in megakaryocytes, interferon α/β signaling has been recently identified as the most significantly downregulated pathway associated with E527K Src hyperactivity.42 One possibility is that persistent activation of tyrosine kinase families in patients with hypermorphic mutations in immune cell signaling results in secondary exhaustion and deactivation of cytokine production, and to the impaired immune responses. This strongly parallels the proposed mechanism of decreased platelet function in patients with GOF Src mutations.
Although the therapeutic management of patients with GOF mutations in BCR signaling pathways remains challenging, potent and selective small molecule inhibitors may emerge as promising therapeutic agents. Besides the most extensively studied JAK inhibitors, compounds targeting other kinases such as SYK, Src-family kinases, or BTK are emerging as new therapeutics for autoimmune diseases.43 If the altered immune phenotype is also found in other pedigrees with SRC-RT, the therapeutic potential of tyrosine kinase inhibitors such as fostamatinib,44 ibrutinib, or rilzabrutinib,45 should be tested.
In summary, in addition to causing defects of platelets and megakaryocytes, SRC-RT may associate with immune dysregulation. Cases of familial ITP should trigger suspicion for this disease, and if confirmed, physicians should attempt an integrated therapeutic approach. The clinical and biological manifestations of a GOF mutation of Src in the described kindred underscore the significant overlap of impaired platelet production and increased platelet consumption and immune dysregulation. Together, this results in a systemic inflammatory state, bridging features of inherited thrombocytopenia and immune thrombosis. Our observations also put forward the possibility of improving patient outcomes by using specific tyrosine kinase inhibitors. Because these patients are very rare, this treatment approach can only be evaluated clinically in an international study.
Acknowledgments
The authors thank Anabel Antón and Angel Esteban-Gil (IMIB-Arrixaca) for assistance with high-throughput sequencing performance and analysis, Raghavendra Palankar for taking and analysing the immunofluorescence images, Belén De la Morena for assistance with Nanopore sequencing, and Francisca Ferrer-Marín and Ernesto Cuenca-Zamora for their help with the Luminex assay. José M. Bastida & José Rivera are the current Coordinators of the Grupo Español de Alteraciones Plaquetarias Congénitas (GEAPC).
This work was partially supported by grants from Instituto de Salud Carlos III (ISCIII) and Feder (PI17/01311, PI17/01966, PI20/00926, and CB15/00055), Fundación Séneca (19873/GERM/15), Gerencia Regional de Salud (GRS 2061A/19 and 2135/A/2020), Fundación Mutua Madrileña (FMM, AP172142019), Sociedad Española de Trombosis y Hemostasia (SETH-FETH; Premio López Borrasca 2019 and Ayuda a Grupos de Trabajo en Patología Hemorrágica), and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), Projektnummer 374031971-TRR 240. The authors’ research on inherited platelet disorders is conducted in accordance with the aims of the Functional and Molecular Characterization of Patients with Inherited Platelet Disorders Project, from Grupo Español de Alteraciones Plaquetarias Congénitas (GEAPC), which is supported by the Spanish Society of Thrombosis and Haemostasis (SETH). V.P.B. has predoctoral contract from IMIB. A.Z.C. holds a PhD fellowship from ISCIII. A.M.Q. holds a PhD fellow form Junta de Castilla y León and Fondo Social Europeo (JCYL- EDU/556/2019 PhD scholarship)
Authorship
Contribution: V.P.-B., A.S.F, A.Z.-C., N.B., J.P., A.M.Q., and J.R. performed most platelet function assays and molecular analysis; C.Z. and A.G. performed and interpreted blood smear immunofluorescence; N.R., A.M.G., J.L.F., A.R.-A., V.V., M.L.L., and J.M.B. performed clinical assessment of patients; V.P.B., N.R., A.G., M.L.L., and J.R. wrote the manuscript; and all authors helped in data interpretation and critically reviewed the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: José Rivera, Centro Regional de Hemodonacion-Universidad de Murcia, Ronda de Garay s/n, Murcia 30003, Spain; e-mail: jose.rivera@carm.es; or María Luisa Lozano, Centro Regional de Hemodonacion-Universidad de Murcia, Ronda de Garay s/n, Murcia 30003, Spain; e-mail: mllozano@um.es.

