

Key Points
RhoB-deficient mice display microthrombocytopenia.
RhoB exerts nonredundant functions in the megakaryocyte lineage compared with RhoA and regulates microtubule dynamics.

Abstract
Megakaryocytes are large cells in the bone marrow that give rise to blood platelets. Platelet biogenesis involves megakaryocyte maturation, the localization of the mature cells in close proximity to bone marrow sinusoids, and the formation of protrusions, which are elongated and shed within the circulation. Rho GTPases play important roles in platelet biogenesis and function. RhoA-deficient mice display macrothrombocytopenia and a striking mislocalization of megakaryocytes into bone marrow sinusoids and a specific defect in G-protein signaling in platelets. However, the role of the closely related protein RhoB in megakaryocytes or platelets remains unknown. In this study, we show that, in contrast to RhoA deficiency, genetic ablation of RhoB in mice results in microthrombocytopenia (decreased platelet count and size). RhoB-deficient platelets displayed mild functional defects predominantly upon induction of the collagen/glycoprotein VI pathway. Megakaryocyte maturation and localization within the bone marrow, as well as actin dynamics, were not affected in the absence of RhoB. However, in vitro–generated proplatelets revealed pronouncedly impaired microtubule organization. Furthermore, RhoB-deficient platelets and megakaryocytes displayed selective defects in microtubule dynamics/stability, correlating with reduced levels of acetylated α-tubulin. Our findings imply that the reduction of this tubulin posttranslational modification results in impaired microtubule dynamics, which might contribute to microthrombocytopenia in RhoB-deficient mice. Importantly, we demonstrate that RhoA and RhoB are localized differently and have selective, nonredundant functions in the megakaryocyte lineage.
Introduction
Blood platelets are small anucleated cells with central roles in hemostasis and thrombosis. Platelets derive from large precursor cells called megakaryocytes (MKs) that are found predominantly in the bone marrow (BM). MK development is characterized by endomitosis (DNA replication without cell division) and cytoplasmic maturation, which comprises the biosynthesis of platelet-specific granules and the formation of an internal demarcation membrane system, which serves as a membrane reservoir for platelet production. Platelet biogenesis is a complex cellular process,1,2 initiated by mature MKs in direct contact with the BM sinusoids,3 and involves the penetration of endothelial cells4,5 and the extension of large cytoplasmic protrusions into the sinusoidal lumen,6,7 which are shed and undergo maturation in the peripheral circulation to become bona fide platelets. The different steps of platelet biogenesis highly depend on rearrangements of both the microtubule (MT) and actin cytoskeletal network.1,2,8,9 Consistently, genetic defects impairing cytoskeletal dynamics can result in thrombocytopenia, as shown in several human disorders and corresponding mouse models.10
Rho GTPases act as molecular switches by cycling between an inactive guanosine diphosphate–bound and an active guanosine triphosphate–bound state. In the latter conformation, Rho GTPases interact with multiple effector proteins to regulate diverse processes including cytoskeletal dynamics, cell adhesion, cell migration, cell polarity, cell cycle, and vesicle trafficking. Multiple members of the Rho family have been established as critical regulators of platelet production and function.11-13 We have previously reported that cytoplasmic MK maturation and subsequent transendothelial platelet biogenesis requires balanced signaling of RhoA and Cdc42.14,15 Mice lacking RhoA in the MK lineage display macrothrombocytopenia (decreased platelet count and increased platelet size), accompanied by a pronounced intravascular mislocalization of whole MKs.15,16
Higher vertebrates express 3 Rho subfamily proteins, RhoA, RhoB, and RhoC, which are 87% homologous on the protein level.17 RhoB is the most divergent member, with a unique C-terminal region containing a variety of lipid modifications,18 which result in its distinct localization to the plasma membrane, Golgi, and endosomes, in contrast to the other 2 members of this subfamily.19,20 In immune cells, RhoB is involved in cytokine trafficking and cell survival21 and affects cell adhesion and migration through β2 and β3 integrins22 but is not required for podosome assembly in macrophages.23 Although the role of RhoB in MKs and platelets remains unknown, its messenger RNA (mRNA) expression has been identified in both cell types. Human MKs, human platelets, and mouse platelets express RhoB mRNA at lower levels than RhoA or RhoC,24,25 whereas in mouse MKs, RhoB shows the highest abundance, followed by RhoA, and comparably lower levels of RhoC.26 The protein levels of RhoB in human platelets were detected at low levels,27 whereas in mouse platelets, it was shown to be more abundant than RhoC.28
In this study, we investigated for the first time the role of RhoB in platelet biogenesis and function by using constitutive knockout mice. We show that, in contrast to RhoA deficiency, loss of RhoB results in microthrombocytopenia, which was accompanied by moderately impaired platelet activation and thrombus formation in vitro. Our results demonstrate that RhoA and RhoB have specific, nonredundant functions in the MK lineage and point to selective defects in MT dynamics in RhoB-deficient mice as a potential mechanism underlying the manifestation of microthrombocytopenia in vivo.
Methods
Mice
Constitutive RhoB-deficient mice (further referred to as RhoB−/−)29 were kindly provided by Ulla G. Knaus. Animal studies were approved by the district government of Lower Franconia (Bezirksregierung Unterfranken).
Statistics
Data presented are mean plus or minus standard deviation (SD). Unpaired, 2-tailed t tests or Mann-Whitney U tests were used to determine statistical significance between 2 groups with normal or nonnormal distribution, respectively. Sample size (n) and statistical significance are reported in the figures or figure legends. Asterisks indicate statistically significant differences compared with wild-type (wt) (*P ≤ .05; **P ≤ .01; ***P ≤ .001). Statistical analysis was performed in GraphPad Prism 9 (GraphPad Software).
Platelet count and size determination, life span, integrin activation, degranulation, glycoprotein expression, aggregation, adhesion under flow conditions, spreading, cold-induced MT disassembly, tail bleeding time, intravital thrombosis model, immunofluorescence microscopy, direct stochastic optical reconstruction microscopy (dSTORM), transmission electron microscopy (TEM) analysis, MK isolation, immunoblotting, and materials are described in detail in supplemental Methods.
Results
RhoB−/− mice display microthrombocytopenia
The consequence of the loss of RhoB in platelets and MKs was studied in constitutive RhoB−/− mice29 and compared with wild-type (wt) littermate controls. The complete loss of RhoB on the protein level was confirmed by immunoblotting in platelets and MKs (Figure 1A; supplemental Figure 1A). The protein levels of other Rho GTPases, namely RhoA, RhoC, Rac1, and Cdc42, remained unaltered in the absence of RhoB, indicating no direct compensatory regulation among these proteins. Using capillary-based quantitative immunoblotting, we found that human platelets expressed lower levels of RhoB protein, which is in agreement with the published proteomics data27,28 (supplemental Figure 1B). Although RhoA-deficient mice displayed a characteristic macrothrombocytopenia,16 loss of RhoB led to microthrombocytopenia (Figure 1B-D). RhoB−/− mice also exhibited a minor decrease in red blood cell counts, suggesting that the protein may play a role in the homeostasis of megakaryocytic and erythroid lineages (supplemental Table 1). The decreased platelet size of RhoB−/− platelets was confirmed by TEM analysis (Figure 1E-F). Resting wt platelets contain an average of 8 to 12 MT coils, known as the marginal band, which are located in the periphery to maintain their spherical shape. Notably, the number of MT coils in RhoB−/− platelets was significantly reduced (wt: 9.48 ± 1.86 vs RhoB−/− 7.43 ± 2.05 MT coils per platelet) (Figure 1E,G). The amount of α- and dense (δ-) granules observed in platelets, on the other hand, was not affected by loss of RhoB (supplemental Figure 1C-D). These results demonstrate that RhoB deficiency affects platelet count and size in vivo.
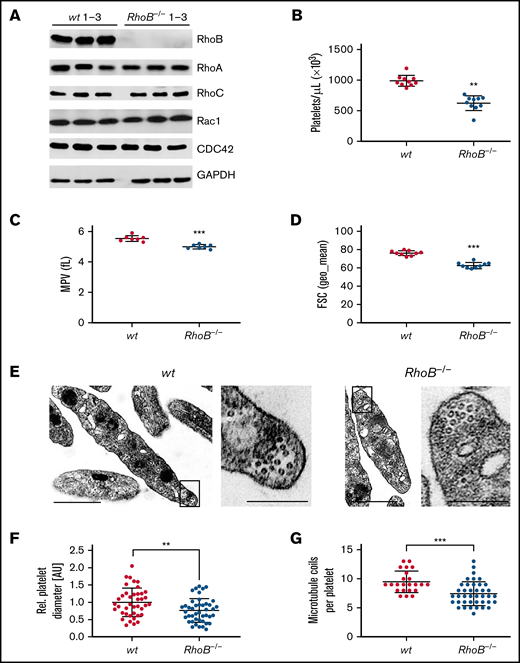
Loss of RhoB leads to microthrombocytopenia and alters microtubule organization in circulating platelets. (A) Blots of platelet lysates from wt and RhoB−/− mice immunoblotted for the indicated proteins. GAPDH was used as loading control (n = 3). (B) Platelet count assessed by flow cytometry. Each data point represents 1 mouse (n = 12). (C) Mean platelet volume measured with a Scilvet blood analyzer. Each data point represents 1 mouse (n = 7). (D) Platelet size assessed by forward scatter characteristics determined by flow cytometry. Each data point represents 1 mouse (n = 12). (E) Representative images of ultrastructural analysis of resting wt (left) and RhoB−/− (right) platelets by transmission electron microscopy (TEM). Scale bar overview, 1 µm; inset, 0.5 µm. (F) Quantification of platelet diameter (relation of platelet width to length) of RhoB−/− platelets relative to wt using TEM images described in panel E (wt n = 4; RhoB−/− n = 5). (G) Quantification of MT coils per platelet using TEM images described in panel E. MT number was determined by manual count of at least 5 imagesper genotype. Each data point represents 1 single platelet (wt n = 4; RhoB−/− n = 5). **P < .01; ***P < .001; Mann-Whitney U test, mean plus or minus SD. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Loss of RhoB leads to microthrombocytopenia and alters microtubule organization in circulating platelets. (A) Blots of platelet lysates from wt and RhoB−/− mice immunoblotted for the indicated proteins. GAPDH was used as loading control (n = 3). (B) Platelet count assessed by flow cytometry. Each data point represents 1 mouse (n = 12). (C) Mean platelet volume measured with a Scilvet blood analyzer. Each data point represents 1 mouse (n = 7). (D) Platelet size assessed by forward scatter characteristics determined by flow cytometry. Each data point represents 1 mouse (n = 12). (E) Representative images of ultrastructural analysis of resting wt (left) and RhoB−/− (right) platelets by transmission electron microscopy (TEM). Scale bar overview, 1 µm; inset, 0.5 µm. (F) Quantification of platelet diameter (relation of platelet width to length) of RhoB−/− platelets relative to wt using TEM images described in panel E (wt n = 4; RhoB−/− n = 5). (G) Quantification of MT coils per platelet using TEM images described in panel E. MT number was determined by manual count of at least 5 imagesper genotype. Each data point represents 1 single platelet (wt n = 4; RhoB−/− n = 5). **P < .01; ***P < .001; Mann-Whitney U test, mean plus or minus SD. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Moderately impaired platelet function in RhoB−/− mice
We next determined the characteristics and function of RhoB−/− platelets in vitro by flow cytometry. Levels of surface glycoproteins (GPs) were not significantly different between RhoB−/− and wt platelets, except for a 13% reduction in GPV and an 18% reduction in GPVI receptor levels (supplemental Table 2), which is likely related to the decreased platelet size. Platelet activation induces a shape change from a resting discoid to a spherical form.30 RhoA-deficient platelets showed selective defects in platelet activation and shape change after stimulation of agonists that induce G-protein–coupled (Gα13, Gαq) signaling pathways, which resulted in impaired intravital thrombosis and a mild bleeding phenotype.16 To investigate whether the absence of RhoB had a similar effect, we analyzed platelet shape change and compared it to RhoA-deficient platelets by light transmission aggregometry upon stimulation with a low dose of the stable thromboxane A2 analog U46619 (Figure 2A). Under these conditions, shape change in RhoB−/− platelets was unaltered, whereas it was abrogated in RhoA-deficient platelets, demonstrating that RhoB is not essential for Gα13 signaling. However, the aggregation of RhoB−/− platelets in response to intermediate concentrations of thrombin, U46619, or convulxin (CVX) was moderately reduced, indicating an overall impairment of platelet function (Figure 2B).
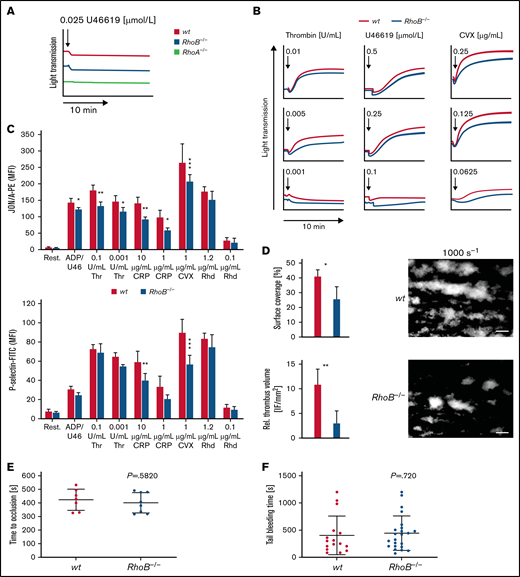
Impaired activation and thrombus formation of RhoB−/− platelets in vitro. (A-B) Platelet light transmission aggregometry. Representative aggregation curves of washed wt and RhoB−/− platelets stimulated with different agonist concentrations under stirring conditions for 10 minutes are shown. (A) Shape change of wt and RhoB−/− platelets in response to low concentrations of the thromboxane analog U46619. RhoA-deficient platelets displaying defective shape change under these conditions were used as a control. (B) Aggregation of RhoB−/− platelets in vitro. (C) Platelet integrin αIIbβ3 activation (JON/A-PE binding; upper) and α-granule secretion (P-selectin-FITC; lower) in vitro were determined upon 6-minute stimulation of wt and RhoB−/− platelets with the indicated agonists at different concentrations (n = 4). (D) In vitro thrombus formation under flow on a collagen-coated surface. Representative images of thrombus formation at an intermediate shear of 1000 s−1 are shown. Upper panel: adhesion of platelets to collagen assessed by area coverage. Lower panel: formation of 3-dimensional aggregates displayed as relative thrombus formation (n = 8). (E) Induction of arterial thrombosis by mechanical injury of the abdominal aorta of wt and RhoB−/− mice in vivo. Each data point represents 1 mouse (wt n = 7; RhoB−/− n = 8). (F) Tail bleeding time on filter paper of wt and RhoB−/− mice in vivo. Each data point represents 1 mouse (wt n = 15; RhoB−/− n = 23. *P < .05; **P < .01; ***P < .001; Mann-Whitney U test, mean plus or minus SD. ADP/U466, adenosine diphosphate/U46619; CRP, collagen-related peptide; CVX, convulxin; Rest, resting; Rhd, rhodocytin; Thr, thrombin.
Impaired activation and thrombus formation of RhoB−/− platelets in vitro. (A-B) Platelet light transmission aggregometry. Representative aggregation curves of washed wt and RhoB−/− platelets stimulated with different agonist concentrations under stirring conditions for 10 minutes are shown. (A) Shape change of wt and RhoB−/− platelets in response to low concentrations of the thromboxane analog U46619. RhoA-deficient platelets displaying defective shape change under these conditions were used as a control. (B) Aggregation of RhoB−/− platelets in vitro. (C) Platelet integrin αIIbβ3 activation (JON/A-PE binding; upper) and α-granule secretion (P-selectin-FITC; lower) in vitro were determined upon 6-minute stimulation of wt and RhoB−/− platelets with the indicated agonists at different concentrations (n = 4). (D) In vitro thrombus formation under flow on a collagen-coated surface. Representative images of thrombus formation at an intermediate shear of 1000 s−1 are shown. Upper panel: adhesion of platelets to collagen assessed by area coverage. Lower panel: formation of 3-dimensional aggregates displayed as relative thrombus formation (n = 8). (E) Induction of arterial thrombosis by mechanical injury of the abdominal aorta of wt and RhoB−/− mice in vivo. Each data point represents 1 mouse (wt n = 7; RhoB−/− n = 8). (F) Tail bleeding time on filter paper of wt and RhoB−/− mice in vivo. Each data point represents 1 mouse (wt n = 15; RhoB−/− n = 23. *P < .05; **P < .01; ***P < .001; Mann-Whitney U test, mean plus or minus SD. ADP/U466, adenosine diphosphate/U46619; CRP, collagen-related peptide; CVX, convulxin; Rest, resting; Rhd, rhodocytin; Thr, thrombin.
To investigate these findings further, we next assessed platelet activation by flow cytometry, which allows for a more detailed analysis of platelet activation. In contrast to the reported selective defect of RhoA-deficient platelets in response to thrombin and U46619,16 RhoB−/− platelets displayed moderately reduced activation when stimulated with most tested agonists. The most profound defect was observed upon stimulation of the collagen/GPVI signaling pathway with either collagen-related peptide (CRP) or CVX (Figure 2C). This was consistent for both integrin αIIbβ3 activation (as assessed by JON/A phycoerythrin [PE] binding31 ) as well as degranulation (as assessed by binding of anti–P-selectin fluorescein isothiocyanate [FITC]). We next measured adenosine triphosphate (ATP) release in lysates of activated platelets using a CellTiter-Glo assay. Although, there was no significant difference between the genotypes; RhoB−/− platelets display a tendency toward reduced ATP release in response to thrombin (supplemental Figure 2A).
It was previously demonstrated that RhoB−/− macrophages displayed reduced level of β2 and β3, but not β1, integrins.23 Because murine platelets do not express β2 integrins, and the surface expression of β3 and β1 integrins was unaltered in RhoB−/− platelets (supplemental Table S2), we investigated platelet β1 integrin activation (as assessed by anti-integrin β1-FITC binding) by flow cytometry and found similar activation defects as for αIIbβ3 integrin activation (supplemental Figure 2B).
We next performed in vitro flow chamber assays, in which anticoagulated whole blood was perfused over a collagen-coated surface at intermediate (1000 s−1) and high shear rates (1700 s−1). RhoB−/− platelets were able to form thrombi under these conditions; however, the overall surface coverage and relative thrombus volume were significantly reduced at intermediate shear compared with wt platelets (Figure 2D; supplemental Figure 2C), indicating that loss of RhoB predominantly affected the GPVI pathway, in accordance with our platelet activation results. To better understand the role of RhoB in GPVI signaling, we performed platelet activation using CVX (1 μg/mL, 90 seconds) in the presence of inhibitors of second-wave mediators followed by immunoblotting using an anti–phospho-tyrosine antibody. Under these conditions, we did not observe major differences in the pattern or intensity of proteins phosphorylated on tyrosine residues (supplemental Figure 2D). Rho-associated protein kinases (ROCK1 and ROCK2), downstream targets of Rho subfamily members, regulate myosin light chain 2 phosphorylation. Myosin light chain 2 phosphorylation was unaltered in platelets in response to thrombin stimulation, indicating that RhoA/RhoC can compensate for the loss of RhoB (supplemental Figure 2E).
The platelet function defects observed in vitro in aggregation, integrin αIIbβ3 activation, P-selectin expression, and thrombus formation studies did not translate into altered arterial thrombus formation in vivo, which was determined by mechanical injury of the abdominal aorta (Figure 2E). Furthermore, unaltered tail bleeding times indicated that hemostasis was not affected in RhoB−/− mice (Figure 2F). These findings stand in contrast to the defects observed in RhoA-deficient mice in vivo and emphasize that RhoA and RhoB are involved in distinct platelet signaling pathways.
Unaltered platelet turnover in RhoB−/− mice
Decreased circulating platelet counts may manifest as a result of impaired platelet generation, an increased platelet clearance rate, or a combination of both. We have previously reported that macrothrombocytopenia in RhoA-deficient mice is associated with increased platelet turnover.16 We therefore investigated the lifespan of circulating, anti–GPIX-labeled platelets over a period of 5 days by flow cytometry (Figure 3A). We observed only a minor reduction of platelet lifespan in RhoB−/− mice, indicating that the reduction in platelet counts was predominantly caused by a direct impairment in platelet production. RhoB−/− mice exhibited splenomegaly; however, the overall spleen morphology and splenic MK numbers were unaltered (Figure 3B-D). Platelet removal was reported to be modulated by desialylation of surface glycoproteins, which, together with macrophage galactose lectin, mediate the clearance of platelets by Kupffer cells.32,33 However, RhoB−/− platelets did not show alterations in Erythrina cristagalli lectin binding to exposed galactose (Figure 3E) and in terminal galactose levels (determined by the ratio of neuraminidase treated platelets vs untreated platelets) (Figure 3F). The amount of RNA-rich, thiazole orange–positive platelets, an indicator of young, reticulated platelets, was not altered in RhoB−/− platelets (Figure 3G). Splenomegaly might be a consequence of the loss of RhoB in other cell types, potentially involving the erythroid lineage, as red blood cell numbers were significantly decreased (supplemental Table 1). Taken together, these results indicate that the thrombocytopenia in RhoB−/− mice was not caused by increased platelet clearance and suggest a direct role for RhoB in either MK maturation or platelet generation.
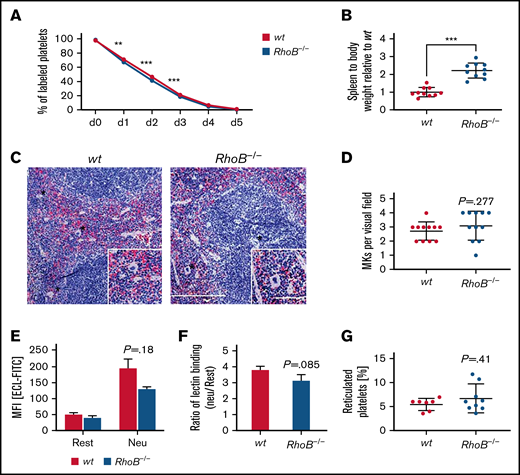
Lack of increased platelet turnover in RhoB−/− mice. (A) For the determination of platelet lifespan, wt and RhoB−/− platelets were labeled with an α-GPIX antibody, and the labeled platelet population was analyzed over 5 consecutive days (n = 5 mice per genotype). Unpaired t test with individual variances computed for each comparison, mean plus or minus SD. (B) Ratio of spleen weight (mg) to body weight (g) of RhoB−/− mice normalized to wt (n = 10 miceper genotype). (C) Paraffin sections of murine spleens stained using hematoxylin and eosin. Analysis was performed at a Leica DMI4000 B microscope. Scale overview, 100 µm; inset, 80 µm. (D) Number of MKs per visual field determined in spleen paraffin sections (n = 3; 4 visual fields per genotype; visual field, 2.3 mm2). (E) Assessment of Erythrina cristagalli lectin–FITC (ECL-FITC) binding to wt and RhoB−/− platelets by flow cytometry at resting state (rest) or upon neuraminidase treatment (Neu) (n = 3). (F) Ratio of lectin binding (neuraminidase-treated platelets vs untreated platelets) for both genotypes (n = 3). (G) Percentage of reticulated (RNA-rich) platelets assessed by thiazole orange binding in flow cytometry (wt n = 7; RhoB−/− n = 9). ***P < .001; Mann-Whitney U test, mean plus or minus SD. MFI, meanfluorescence intensity.
Lack of increased platelet turnover in RhoB−/− mice. (A) For the determination of platelet lifespan, wt and RhoB−/− platelets were labeled with an α-GPIX antibody, and the labeled platelet population was analyzed over 5 consecutive days (n = 5 mice per genotype). Unpaired t test with individual variances computed for each comparison, mean plus or minus SD. (B) Ratio of spleen weight (mg) to body weight (g) of RhoB−/− mice normalized to wt (n = 10 miceper genotype). (C) Paraffin sections of murine spleens stained using hematoxylin and eosin. Analysis was performed at a Leica DMI4000 B microscope. Scale overview, 100 µm; inset, 80 µm. (D) Number of MKs per visual field determined in spleen paraffin sections (n = 3; 4 visual fields per genotype; visual field, 2.3 mm2). (E) Assessment of Erythrina cristagalli lectin–FITC (ECL-FITC) binding to wt and RhoB−/− platelets by flow cytometry at resting state (rest) or upon neuraminidase treatment (Neu) (n = 3). (F) Ratio of lectin binding (neuraminidase-treated platelets vs untreated platelets) for both genotypes (n = 3). (G) Percentage of reticulated (RNA-rich) platelets assessed by thiazole orange binding in flow cytometry (wt n = 7; RhoB−/− n = 9). ***P < .001; Mann-Whitney U test, mean plus or minus SD. MFI, meanfluorescence intensity.
RhoB shows a distinct localization in megakaryocytes compared with RhoA
To better understand its role in MKs, we investigated the localization of RhoB in in vitro–differentiated mouse MKs and compared it to RhoA using immunofluorescence microscopy. We found a very distinct plasma membrane–associated staining for RhoB and a more diffuse, cytoplasmic staining for RhoA (Figure 4A), which is in agreement with their subcellular localization in other cells.18 Next, we stained healthy human bone marrow in situ for RhoB and found a similar staining pattern (Figure 4B).
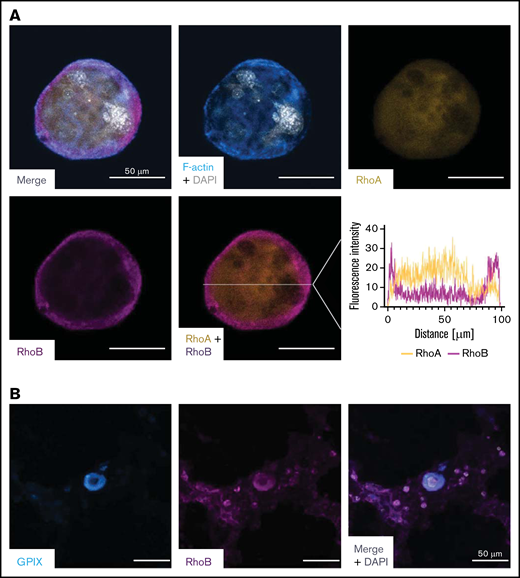
Staining of RhoA and RhoB in murine and human MKs. (A) Representative confocal images of in vitro–differentiated MKs 24 hours after bovine serum albumin gradient. The MKs were spun down to Poly-L-Lysine–coated glass slides and stained with Phallodin-Alexa488 (cyan), RhoA-Alexa546 (yellow), RhoB-Alexa647 (magenta), and DAPI to visualize the nucleus. Scale bar, 50 µm. Localization of RhoA and RhoB was analyzed by measuring the signal intensity on the white line (n = 2). (B) Representative confocal image of the human BM. Cryosections were stained with GPIX-FITC, RhoB-A546, and DAPI to visualize the nucleus. Scalebar, 50 µm. DAPI, 4′,6-diamidino-2-phenylindole.
Staining of RhoA and RhoB in murine and human MKs. (A) Representative confocal images of in vitro–differentiated MKs 24 hours after bovine serum albumin gradient. The MKs were spun down to Poly-L-Lysine–coated glass slides and stained with Phallodin-Alexa488 (cyan), RhoA-Alexa546 (yellow), RhoB-Alexa647 (magenta), and DAPI to visualize the nucleus. Scale bar, 50 µm. Localization of RhoA and RhoB was analyzed by measuring the signal intensity on the white line (n = 2). (B) Representative confocal image of the human BM. Cryosections were stained with GPIX-FITC, RhoB-A546, and DAPI to visualize the nucleus. Scalebar, 50 µm. DAPI, 4′,6-diamidino-2-phenylindole.
Impaired microtubule organization and proplatelet formation in RhoB−/− MKs
We next investigated the outcome of RhoB deficiency on MK maturation and function. MK numbers in the BM compartment were unaltered (Figure 5A). Investigation of the ultrastructure of BM MKs by TEM in situ revealed a normally developed demarcation membrane system and granules in RhoB−/− MKs (Figure 5B). In situ analysis of BM cryosections stained for CD105 (vessel marker) and GPIX (MK marker) revealed that RhoB had no influence on MK localization near the BM vasculature with sinusoidal contact (Figure 5C-D). Together, these results indicated that RhoB is dispensable for MK maturation and localization in vivo. This is in marked contrast to the intrasinusoidal MK mislocalization observed in RhoA-deficient mice, which again emphasizes the differential functions of RhoB and RhoA not only in platelets but also in MKs.
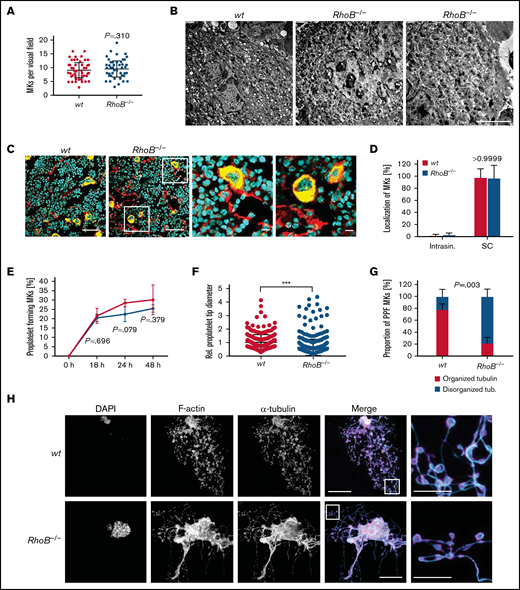
Decreased proplatelet tip size and impaired microtubule organization of RhoB−/− MKs in vitro. (A) Quantification of MK numbers in wt and RhoB−/− cryosections of the fluorescently labeled BM. At least 10 fluorescence images/mouse were analyzed using a confocal microscope. Each data point represents 1 MK (n = 7). Unpaired, 2-tailed t test, mean plus or minus SD. (B) Representative images of ultrastructural analysis of wt and RhoB−/− MKs in situ using TEM. Scale bar, 4 µm. (C) Representative confocal microscopy images of femur cryosections. The femora were processed using the Kawamoto method for cryosections. Sections were stained with fluorescently labeled anti-CD105 antibody to stain vessels (red), and fluorescently labeled anti-GPIX antibody to visualize MKs (yellow) and DAPI (blue). Scale bar in overview, 40 µm; in zoom, 5 µm. (D) Quantification of MK localization in wt and RhoB−/− BM in images shown in panel B. (E-H) Analysis of proplatelet formation of BM-derived RhoB−/− MKs. (E) Quantification of in vitro proplatelet formation at 18 hours, 24 hours, and 48 hours after enrichment by bovine serum albumin (BSA) gradient. Graph shows percentage of MKs that perform PPF (n = 3 mice per genotype). Multiple unpaired 2-tailed Student t test with Holm-Sidak correction for multiple comparisons, mean plus or minus SD. (F) Quantification of proplatelet tip diameter of wt and RhoB−/− MKs 24 hours after BSA gradient (normalized to wt, n = 2 mice per genotype). At least 11 MKs per mice were analyzed. Each data point represents 1 measured proplatelet tip (wt = 250; RhoB−/− = 494). ***P < .001; Mann-Whitney U test, mean plus or minus SD. (G) Quantification of MT organization in enriched wt and RhoB−/− MKs (n = 3 mice per genotype). At least 13 images/mouse were analyzed. Mann-Whitney U test, mean plus or minus SD. (H) Representative confocal images of in vitro–differentiated MKs 24 hours after BSA gradient. The proplatelets were spun down to Poly-L-Lysine–coated glass slides and stained with phalloidin-Atto647 (magenta), α-tubulin-Alexa488 (cyan), and DAPI to visualize the nucleus. (n = 5) Scale bar in overview, 40 µm; inset, 5 µm. DAPI, 4′,6-diamidino-2-phenylindole; PPF, proplatelet-forming; Intrasin., intrasinusoidal; SC, sinusoidal contact; TB, tubulin.
Decreased proplatelet tip size and impaired microtubule organization of RhoB−/− MKs in vitro. (A) Quantification of MK numbers in wt and RhoB−/− cryosections of the fluorescently labeled BM. At least 10 fluorescence images/mouse were analyzed using a confocal microscope. Each data point represents 1 MK (n = 7). Unpaired, 2-tailed t test, mean plus or minus SD. (B) Representative images of ultrastructural analysis of wt and RhoB−/− MKs in situ using TEM. Scale bar, 4 µm. (C) Representative confocal microscopy images of femur cryosections. The femora were processed using the Kawamoto method for cryosections. Sections were stained with fluorescently labeled anti-CD105 antibody to stain vessels (red), and fluorescently labeled anti-GPIX antibody to visualize MKs (yellow) and DAPI (blue). Scale bar in overview, 40 µm; in zoom, 5 µm. (D) Quantification of MK localization in wt and RhoB−/− BM in images shown in panel B. (E-H) Analysis of proplatelet formation of BM-derived RhoB−/− MKs. (E) Quantification of in vitro proplatelet formation at 18 hours, 24 hours, and 48 hours after enrichment by bovine serum albumin (BSA) gradient. Graph shows percentage of MKs that perform PPF (n = 3 mice per genotype). Multiple unpaired 2-tailed Student t test with Holm-Sidak correction for multiple comparisons, mean plus or minus SD. (F) Quantification of proplatelet tip diameter of wt and RhoB−/− MKs 24 hours after BSA gradient (normalized to wt, n = 2 mice per genotype). At least 11 MKs per mice were analyzed. Each data point represents 1 measured proplatelet tip (wt = 250; RhoB−/− = 494). ***P < .001; Mann-Whitney U test, mean plus or minus SD. (G) Quantification of MT organization in enriched wt and RhoB−/− MKs (n = 3 mice per genotype). At least 13 images/mouse were analyzed. Mann-Whitney U test, mean plus or minus SD. (H) Representative confocal images of in vitro–differentiated MKs 24 hours after BSA gradient. The proplatelets were spun down to Poly-L-Lysine–coated glass slides and stained with phalloidin-Atto647 (magenta), α-tubulin-Alexa488 (cyan), and DAPI to visualize the nucleus. (n = 5) Scale bar in overview, 40 µm; inset, 5 µm. DAPI, 4′,6-diamidino-2-phenylindole; PPF, proplatelet-forming; Intrasin., intrasinusoidal; SC, sinusoidal contact; TB, tubulin.
We next analyzed the ability of RhoB−/− MKs to produce proplatelets in vitro. For this, BM-derived MKs were cultured for 3 days in the presence of thrombopoietin and hirudin to induce proplatelet formation,34 followed by staining for F-actin and α-tubulin (Figure 5E-H). We could not detect alterations in the kinetics of proplatelet formation between enriched wt and RhoB−/− MKs during the observation time of 48 hours (Figure 5E), indicating that the ability to fragment into proplatelets was still preserved in the absence of RhoB. However, we observed an overall decreased proplatelet tip size and simultaneous unaffected F-actin distribution (Figure 5F,H; supplemental Figure 3A). Strikingly, a high proportion (78.1%) of proplatelets formed by RhoB−/− MKs displayed elongated irregular shaped proplatelet tips with a less uniform tubulin staining and/or enlarged proplatelet shafts compared with the wt, indicating a disorganized MT network in these cells (Figure 5G-H; supplemental Figure 3A).
To assess the functionality of F-actin dynamics in the absence of RhoB, we additionally analyzed the characteristic integrin- and actin-dependent formation of podosomes in in vitro–differentiated MKs upon adhesion on collagen-coated cover slips. Importantly, podosome formation along collagen fibers, as well as the overall signal intensity of F-actin, was similar in wt and RhoB−/− MKs (supplemental Figure 3B-D). Consistently, and in contrast to observations of MKs with impaired podosome formation (eg, upon Cdc42 or Profilin-1 deficiency),14,35 loss of RhoB did not affect the total MK spreading area (supplemental Figure 3E). The distribution of tubulin was also not altered between wt and RhoB−/− MKs under these adhesive conditions (supplemental Figure 3F), pointing to a specific requirement of RhoB in the regulation of MT dynamics during proplatelet formation. Collectively, these results demonstrate that RhoB is a critical modulator of MT, but not actin dynamics, in MKs during proplatelet formation, which might provide an explanation for the decreased size of circulating RhoB−/− platelets in vivo.
RhoB is a critical regulator of microtubule stability in MKs and platelets
To investigate the mechanism underlying the tubulin organization defect during proplatelet formation upon loss of RhoB, we investigated cytoskeletal regulators in MKs and platelets. Total protein levels of α-tubulin, and β1-tubulin, which is exclusively expressed in the MK lineage, were unaltered in BM-derived in vitro–differentiated MKs and platelets (supplemental Figure 4A). Protein levels of downstream effectors of the Rho subfamily proteins, such as nonmuscle myosin IIA and IIB, that influence cell migration and contractility,36 or the formins mDia1 and mDia2, which orchestrate actin and microtubule remodeling during PPF,37,38 were also unchanged (supplemental Figure 4B-C). Levels of the podosome-associated proteins vinculin and Arp239 were not affected, in line with functional podosome formation in RhoB−/− MKs (supplemental Figure 4B). The levels of the MT plus-end tracking proteins end-binding protein 3 and adenomatous polyposis coli (APC), which was recently reported as a negative regulator of proplatelet formation in mice,40 were also unchanged in RhoB−/− MKs (supplemental Figure 4A,C).
These findings indicated that RhoB may directly influence MT dynamics during platelet formation. Due to the heterogeneity of MK cultures, we decided to focus on the circulating platelets (ie, the terminal in vivo products of proplatelet formation). We analyzed MT and F-actin distribution in adherent, spread platelets in vitro. For this, platelets were incubated for 5, 15, and 30 minutes on fibrinogen-coated cover slips, which induces extensive spreading of wt platelets that is supposed to be driven by integrin outside-in signaling. Similar to the normal adhesion and spreading of MKs on collagen, we did not observe a difference in the spreading kinetics between wt and RhoB−/− platelets (Figure 6A-B), indicating that integrin outside-in signaling is functional in the absence of RhoB.
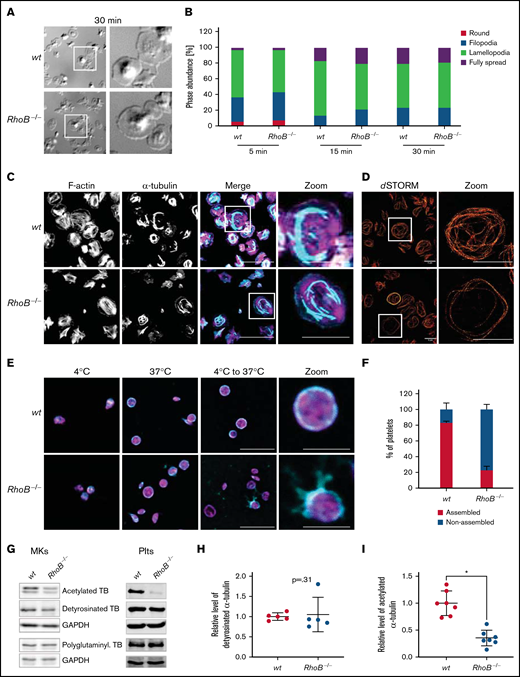
Impaired microtubule stability in RhoB−/− platelets. (A-B) Spreading and F-actin dynamics in RhoB−/− platelets. (A) Representative images of platelets spread on fibrinogen-coated cover slips in the presence of 0.01 U/mL thrombin for 30 minutes. (B) Quantification of platelet spreading on fibrinogen for 5, 10, and 15 minutes (n = 3). *P < .05; Mann-Whitney U test, mean plus or minus SD. (C-D) Analysis of spread platelets on fibrinogen by immunofluorescence confocal and superresolution (dSTORM) microscopy. (C) Representative confocal microscopy images of platelets spread on fibrinogen for 30 minutes with 0.01 U/mL thrombin, stained with phalloidin-Atto647 (magenta) and α-tubulin-Alexa488 (cyan), and imaged using a Leica TC SP8 confocal microscope. Scale bar in overview, 10 µm; scale bar in zoom, 5 µm (n = 4). (D) Representative dSTORM images of platelets spread on fibrinogen for 30 minutes, stained with α-tubulin-Alexa488 (red), and imaged using an in-house-made dSTORM microscope. Scale bar in overview and zoom, 5 µm (n = 2). (E-F) Analysis of cold-induced MT disassembly and reassembly of spread platelets in vitro. (E) Representative confocal immunofluorescence microscopy images of MT organization at 4°C and 37°C and a combination of disassembly at 4°C with following reassembly at 37°C in platelets. Platelets were stained with phalloidin-Atto647 (magenta) and α-tubulin-Alexa488 (cyan). Scale bar in overview, 10 µm; zoom, 3 µm (n = 3). (F) Quantification of MT organization in wt and RhoB−/− platelets after cold-induced MT disassembly and reassembly (4°C to 37°C). At least 5 images/mouse were analyzed, with minimum of 40 platelets per genotype (n = 3). *P < .05; Mann-Whitney U test, mean plus or minus SD. (G-I) Analysis of acetylation and detyrosination and polyglutaminylation of α-tubulin residues by immunoblotting. (G) Platelet and MK lysates of wt and RhoB−/− mice were immunoblotted for posttranslational modifications (PTM) of α-tubulin(acetylated α-tubulin, detyrosinated α-tubulin, and polyglutaminylated α-tubulin). (H-I) Quantification of PTM immunoblots. Each data point represents 1 mouse (acetylated α-tubulin n = 7; glutaminylated α-tubulin n = 5). *P < .05; Mann-Whitney U test, mean plus or minus SD. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; TB, tubulin.
Impaired microtubule stability in RhoB−/− platelets. (A-B) Spreading and F-actin dynamics in RhoB−/− platelets. (A) Representative images of platelets spread on fibrinogen-coated cover slips in the presence of 0.01 U/mL thrombin for 30 minutes. (B) Quantification of platelet spreading on fibrinogen for 5, 10, and 15 minutes (n = 3). *P < .05; Mann-Whitney U test, mean plus or minus SD. (C-D) Analysis of spread platelets on fibrinogen by immunofluorescence confocal and superresolution (dSTORM) microscopy. (C) Representative confocal microscopy images of platelets spread on fibrinogen for 30 minutes with 0.01 U/mL thrombin, stained with phalloidin-Atto647 (magenta) and α-tubulin-Alexa488 (cyan), and imaged using a Leica TC SP8 confocal microscope. Scale bar in overview, 10 µm; scale bar in zoom, 5 µm (n = 4). (D) Representative dSTORM images of platelets spread on fibrinogen for 30 minutes, stained with α-tubulin-Alexa488 (red), and imaged using an in-house-made dSTORM microscope. Scale bar in overview and zoom, 5 µm (n = 2). (E-F) Analysis of cold-induced MT disassembly and reassembly of spread platelets in vitro. (E) Representative confocal immunofluorescence microscopy images of MT organization at 4°C and 37°C and a combination of disassembly at 4°C with following reassembly at 37°C in platelets. Platelets were stained with phalloidin-Atto647 (magenta) and α-tubulin-Alexa488 (cyan). Scale bar in overview, 10 µm; zoom, 3 µm (n = 3). (F) Quantification of MT organization in wt and RhoB−/− platelets after cold-induced MT disassembly and reassembly (4°C to 37°C). At least 5 images/mouse were analyzed, with minimum of 40 platelets per genotype (n = 3). *P < .05; Mann-Whitney U test, mean plus or minus SD. (G-I) Analysis of acetylation and detyrosination and polyglutaminylation of α-tubulin residues by immunoblotting. (G) Platelet and MK lysates of wt and RhoB−/− mice were immunoblotted for posttranslational modifications (PTM) of α-tubulin(acetylated α-tubulin, detyrosinated α-tubulin, and polyglutaminylated α-tubulin). (H-I) Quantification of PTM immunoblots. Each data point represents 1 mouse (acetylated α-tubulin n = 7; glutaminylated α-tubulin n = 5). *P < .05; Mann-Whitney U test, mean plus or minus SD. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; TB, tubulin.
To analyze the distribution of F-actin and MTs, spread platelets were stained for α-tubulin and F-actin. In line with results from MKs, the distribution of F-actin was not affected by RhoB deficiency. Consistently, F-actin content and assembly upon CRP or thrombin stimulation were unaltered in RhoB−/− platelets (supplemental Figure 5A), emphasizing that RhoB is dispensable for F-actin dynamics in MKs and platelets. In contrast, the α-tubulin network appeared markedly altered in RhoB−/− platelets with shorter MT filament length (Figure 6C). We therefore next visualized the MT network in spread platelets by superresolution microscopy (dSTORM) as described previously.41 Indeed, we observed a high proportion of RhoB−/− platelets with fewer peripheral MT coils and an overall less dense MT network compared with the wt (Figure 6D; supplemental Figure 5B). These results were in line with the decreased number of tubulin coils in resting RhoB−/− platelets (Figure 1E,G) and pointed to a critical role of RhoB in MT dynamics.
MTs are highly dynamic structures that are constantly assembled and disassembled. MT disassembly into dimers can be artificially induced by exposure to cold (4°C), and reassembly into fibers occurs spontaneously at 37°C.42,43 Although both wt and RhoB−/− platelets displayed MT coils at their periphery at 37°C, which disassembled upon cold storage, only RhoB−/− platelets were not able to reassemble MT coils after disassembly (wt: 83 ± 2% vs RhoB−/−: 23 ± 5%) (Figure 6E-F; supplemental Figure 5C).
MTs can be modified through PTMs, which serve the adaption to different cellular functions.44 In platelets, PTMs of α-tubulin have been reported to be involved in MT rearrangements, including acetylation of residue K40 and detyrosination/tyrosination (in C-terminal tyrosines, often also called glutaminylation).45 Acetylation and detyrosination are indicators for stable, longer-lived MTs, whereas more dynamic MTs are tyrosinated and deacetylated.44 We therefore investigated the acetylation status of the α-tubulin K40 residue by immunoblotting. RhoB−/− platelets and MKs showed pronouncedly decreased levels of acetylated α-tubulin (Figure 6G-I), which was by tendency also observed in MK lysates. In contrast, levels of detyrosinated and polyglutaminylated α-tubulin were unaltered in both platelets and MKs (Figure 6G-H). Thus, deficiency in RhoB results in short-lived MTs that are either per se more unstable or constantly remodeled. Reduced acetylation levels were not due to a change in levels of a major deacetylation enzyme, HDAC6 (supplemental Figure 4C).
In summary, these results reveal that RhoB has a nonredundant role as an important regulator of MT turnover or stability and suggest that the impaired MT dynamics contribute to defective platelet biogenesis and microthrombocytopenia in RhoB−/− mice.
Discussion
Here, we show that, in contrast to MK-specific deficiency of RhoA or Cdc42,16,46 loss of the small Rho GTPase RhoB results in microthrombocytopenia in mice, which might involve a profound defect in MT stability.
The majority of platelet disorders associated with mutations in cytoskeletal genes are characterized by macrothrombocytopenia, which is reflected in transgenic mouse models of the respective diseases.10 Microthrombocytopenia, on the other hand, is a rare clinical condition. It is a characteristic of the Wiskott-Aldrich syndrome (WAS) and X-linked thrombocytopenia, both of which are caused by mutations in the WAS gene,47,48 as well as congenital autosomal-recessive small-platelet thrombocytopenia caused by mutations in adhesion and degranulation-promoting adaptor protein (ADAP).49,50 Both WAS protein (WASP) and ADAP are primarily known as regulators of the actin cytoskeleton: ADAP is a scaffolding protein involved in receptor-induced actin cytoskeletal dynamics in platelets.41 WASP is a downstream effector of Cdc42 and, via the Arp2/3 complex, leads to actin cytoskeletal rearrangements associated with filopodia formation. In line with this pathway, loss of Arp2 in MKs in mice,51 or mutations in the Arp2/3 complex component ARPC1B in humans, leads to microthrombocytopenia.52 Interestingly, deficiency of Profilin-1, another protein associated with actin turnover, in MKs results in a WAS-like phenotype in mice.35
Profilin-1 and Arp2 deficiency is linked to altered MT stability,35,51 which resembles our observations on the RhoB−/− mice. However, there are 2 striking differences: first, a major characteristic of mouse models with WAS- or ADAP-like phenotypes is the release of proplatelets into the BM compartment, referred to as “ectopic platelet release.”35,41,48,51 Second, platelets and MKs lacking ADAP, Arp2, or WASP display pronounced defects in F-actin dynamics, including aberrant podosome formation. In contrast, RhoB−/− MKs do not show signs of ectopic platelet release, and our results indicate largely normal F-actin organization, including podosome formation in vitro. Furthermore, protein levels of Profilin-1 and Arp2 are unaffected in the absence of RhoB. Together, our findings point to impaired MT, but not actin, dynamics as a potential mechanism underlying the manifestation of microthrombocytopenia in vivo. Our results suggest that the characteristic phenotype of ectopic platelet release is critically dependent on aberrant actin dynamics upon dysregulation of the WASP/Arp2/3 pathway.
In this study, we provide evidence that in MKs and platelets, RhoB and RhoA have very distinct functions. RhoA-deficient mice display largely normal MK maturation; however, a transmigration of entire MKs into BM sinusoids contributes tomacrothrombocytopenia.16 The phenotypic differences might be explained by the unique subcellular localization of RhoB compared with RhoA.21 Although RhoB was specifically localized close to the plasma membrane, RhoA was more evenly distributedthroughout the cytoplasm of MKs. Although we only found low levels of RhoB in human platelets, studies using transcriptomic approaches identified RhoB mRNA in human MKs,25 which we were able to confirm on the protein level (Figure 4). In murine MKs, we identified RhoB mRNA as most abundantly expressed among the Rho subfamily members.26
In contrast to the selective Gα13/Gαq-signaling defect in RhoA-deficient platelets, RhoB−/− platelets showed an impaired platelet activation in vitro independent of the agonist used, but most pronounced upon stimulation of the collagen receptor GPVI, which may partially be explained by the 18% reduction in GPVI receptor levels. Upon inhibition of second-wave mediators, GPVI signaling was not altered, suggesting that reduced thrombus formation in vitro might be a consequence of reduced signaling downstream of G-protein-coupled receptors (activated by adenosine diphosphate or thromboxane A2). This is supported by the lower tendency of ATP secretion in RhoB−/− platelets and together could point to a reduction in the feedback activation as a potential reason underlying the reduced response of platelets to GPVI agonists. Despite the differences observed in platelet activation, aggregation, and thrombus formation in vitro, arterial thrombus formation and bleeding time were not altered in the absence of RhoB, highlighting that additional signaling pathways can fully compensate the reduced activation of platelets in response to GPVI stimulation, similar to what has been described in Grb2-deficient mice.53
Interestingly, RhoB activity was shown to regulate Rac1 translocation to endosomes in endothelial cells.54 Because Rac1 is a critical regulator of PLCγ2 activation downstream of GPVI in platelets,55,56 loss of RhoB may affect Rac1 localization and function in platelets, causing predominantly GPVI-related signaling defects. However, in contrast to Rac1-deficiency, lamellipodia formation was not affected in the absence of RhoB.
RhoB has mostly been described as a regulator of actin dynamics in diverse cell types where it, similar to RhoA, regulates stress fiber formation57 and cell migration.18,21 Interestingly, the F-actin distribution of RhoB−/− platelets and MKs appeared unaltered, which may indicate that RhoA is able to compensate for the loss of RhoB in regulating actin dynamics in the MK lineage. In contrast, RhoB−/− platelets displayed a pronounced MT assembly defect, evident by the inability to reassemble MT coils after cold storage, altered α-tubulin distribution in spread platelets, and decreased numbers of MT coils in resting platelets. MT dynamics are critical for the formation of proplatelets during the later stages of thrombopoiesis in vitro and in vivo. Consistently, and in line with our observations in platelets, the MT organization of RhoB−/− proplatelets was profoundly impaired, resulting in aberrantly sized and overall smaller proplatelet tips. These results thus provide a potential explanation for the decreased size of circulating platelets in RhoB−/− mice. This stands in contrast to the phenotype of Tubb1−/− mice, in which the complete loss of the tubulin β1 results in a pronounced macrothrombocytopenia.58 These findings imply that altered MT regulation can have different consequences compared with tubulin deficiency.
Mechanistically, our results indicate that the marked inability to acetylate α-tubulin, a posttranslational modification associated with stable, long-lived MTs, contributes to their decreased stability in RhoB−/− platelets and MKs. Previous studies on the platelet marginal band have shown that α-tubulin is heavily acetylated in platelets and contributes to the kinetics of platelet spreading.59,60 Furthermore, acetylation levels of K40 of α-tubulin are associated with increased tubulin longevity,44 suggesting that decreased acetylation levels might correspond to either direct MT instability or increased MT turnover. This may explain the shortened MT coils in spread RhoB−/− platelets. Moreover, the finding that RhoB−/− platelets are not able to rebuild MT coils after cold-induced disassembly suggests that the formed MT coils are more susceptible to stress.
The signaling pathways that link RhoB to tubulin acetylation remain to be defined in future studies. The tubulin-lysine deacetlyase HDAC6 or the acetyltransferase ATAT1 are obvious candidates,61 which could potentially be involved in controlling acetylation levels downstream of RhoB, although HDAC6 levels were unchanged in platelets or MKs. Loss of the MT plus-end tracking protein APC in MKs and platelets resulted in decreased α-tubulin acetylation at K40.40 However, protein levels of APC were unaltered in RhoB−/− MKs. Additional target proteins linking RhoB to the MT cytoskeleton could be twinfilin 1 (twf1) and cofilin1, which were recently shown as modulators of the actin/tubulin crosstalk during platelet biogenesis.62 Other PTMs of tubulin with important roles in platelets and MKs,9 including detyrosination and polyglutaminylation,63 were also unaltered, indicating a specific role for RhoB in regulating tubulin acetylation.
In summary, our results reveal that impaired MT dynamics, in combination with normal cytoplasmic MK maturation, might contribute to microthrombocytopenia in RhoB−/− mice. Although genetic RhoB deficiency in humans has not been described to date, mutations resulting in persistent RhoB activation were shown to result in systemic capillary leak syndrome64 and an increased risk of cerebral palsy.65 The analysis of platelets from these patients may provide new insights into the roles of RhoB in the MK lineage in humans.
Acknowledgments
The authors thank Stefanie Hartmann, Sylvia Hengst, Birgit Midloch, and Daniela Naumann for excellent technical assistance; Lukas J. Weiss for his help in obtaining anonymized residual human bone marrow samples; Katharina Remer for support with documentation of animal experimentation; and the microscopy platform of the Core Unit Imaging, Rudolf Virchow Center for providing technical infrastructure and support. The graphical abstract was created with Biorender.com.
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) (project number 374031971-TRR 240/project A01 to I.P. and B.N., and project A03 to H.S.). Z.N. was supported by a grant of the German Excellence Initiative to the Graduate School of Life Sciences (GSLS), University of Würzburg. The JEOL JEM-2100 transmission electron microscope was funded by the DFG (project number 218894163).
Authorship
Contribution: M.E., K.A., B.N., I.P., and Z.N., conceptualization; M.E., K.A., I.C.B., A.G., T.H., L.M.W., C.K., K.M., G.H.M.A., A.A.B., S.D., H.S., I.P., and Z.N., investigation; U.G.K., C.S., H.S., B.N., and I.P., resources; B.N., I.P., and Z.N., supervision; : M.E., K.A., B.N., I.P., and Z.N., writing, original draft; and M.E., I.C.B., K.M., U.G.K., C.S., H.S., B.N., I.P., and Z.N., writing, review, and editing.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Zoltan Nagy, Institute of Experimental Biomedicine, University Hospital and Rudolf Virchow Center, University of Würzburg, Josef-Schneider-Straße 2, 97080 Würzburg, Germany; e-mail: nagy_z@ukw.de; and Bernhard Nieswandt, Institute of Experimental Biomedicine, University Hospital and Rudolf Virchow Center, University of Würzburg, Josef-Schneider-Straße 2, 97080 Würzburg, Germany; e-mail: bernhard.nieswandt@virchow.uni-wuerzburg.de.

