

Key Points
In a contemporary cohort of 444 young MPN patients, risks of thrombosis, hemorrhage, and transformation were 1% pt/y.
Current risk scores had no utility. Uniquely, we identify that splenomegaly and hyperviscosity symptoms predict thrombosis and transformation.

Abstract
Myeloproliferative neoplasms (MPNs) are uncommon in children/young adults. Here, we present data on unselected patients diagnosed before 25 years of age included from 38 centers in 15 countries. Sequential patients were included. We identified 444 patients, with median follow-up 9.7 years (0-47.8). Forty-nine (11.1%) had a history of thrombosis at diagnosis, 49 new thrombotic events were recorded (1.16% patient per year [pt/y]), perihepatic vein thromboses were most frequent (47.6% venous events), and logistic regression identified JAK2V617F mutation (P = .016) and hyperviscosity symptoms (visual disturbances, dizziness, vertigo, headache) as risk factors (P = .040). New hemorrhagic events occurred in 44 patients (9.9%, 1.04% pt/y). Disease transformation occurred in 48 patients (10.9%, 1.13% pt/y), usually to myelofibrosis (7.5%) with splenomegaly as a novel risk factor for transformation in essential thrombocythemia (ET) (P= .000) in logistical regression. Eight deaths (1.8%) were recorded, 3 after allogeneic stem cell transplantation. Concerning conventional risk scores: International Prognostic Score for Essential Thrombocythemia-Thrombosis and new International Prognostic Score for Essential Thrombocythemia-Thrombosis differentiated ET patients in terms of thrombotic risk. Both scores identified high-risk patients with the same median thrombosis-free survival of 28.5 years. No contemporary scores were able to predict survival for young ET or polycythemia vera patients. Our data represents the largest real-world study of MPN patients age < 25 years at diagnosis. Rates of thrombotic events and transformation were higher than expected compared with the previous literature. Our study provides new and reliable information as a basis for prospective studies, trials, and development of harmonized international guidelines for the specific management of young patients with MPN.
Introduction
Myeloproliferative neoplasms (MPNs) are clonal myeloid disorders commonest in patients over 60 years with associated risks of thrombosis, hemorrhage, evolution to secondary myelofibrosis (SMF), accelerated phase (AP), and acute myeloid leukemia (AML).1,2
According to current guidelines3,4 treatments are adapted to risk classification based on age and history of thrombosis mainly to reduce the risk of thrombosis and hemorrhage.3 But are based upon literature mostly comprised of patients >60 years. Cohorts of young patients with MPN, defined variably as <60 or <40 years old, have been published5-8 ; sparse data are available concerning patients aged <25 years at diagnosis.
We recently published a literature review focusing on published data for young patients diagnosed ≤20 years,9 471 patients (396 essential thrombocythemia [ET], 75 polycythemia vera [PV]), recording infrequent postdiagnosis thromboses (9.3% PV, 3.8% ET), hemorrhage (4%, 4.8%, respectively), and evolution into SMF (2.7% and 1.7%). Young MPN patients are considered at very low risk although not exempt from complications. Publications focusing on primary myelofibrosis (PMF) are also available.10-13
To broaden knowledge about contemporary young MPN patients and avoid pitfalls of publication bias, we launched a retrospective study among members of the European Hematology Association (EHA) MPN scientific working group (SWG).
Methods
Patient recruitment
Members of the EHA MPN SWG included sequential patients with a diagnosis of MPN before the age of 25 years, excluding suspected/confirmed hereditary cases. Local approval, including institutional review board where required, was gained. The study was conducted in accordance with the Declaration of Helsinki.
Patient characteristics
At diagnosis, the following were recorded: age, sex, mode of presentation, symptoms, history of relevant events, familial history of hemopathies, biological data including blood counts, driver mutation, and wider molecular status and information from bone marrow assessment if available. Diagnosis of MPN was made in accordance with concomitant criteria: PV, ET, PMF, prefibrotic myelofibrosis (PreMF), and unclassified MPN (MPN-u).4,14,15 Of note, histopathologists used adult MPN criteria to diagnose children and adolescent MPNs as no specific guidelines exist for this population. No central diagnostic review was performed.
We assessed the motive for and type of treatments, venesection, antithrombotic, and cytoreductive drugs. Predefined endpoints were collected. Outcomes were assessed using established international prognostic scores for survival and thrombosis, specifically International Prognostic Score for Essential Thrombocythemia-Thrombosis (IPSET-T), new International Prognostic Score for Essential Thrombocythemia-Thrombosis score (IPSET-NT), IPSET survival score (ET patients), PV survival score (PV patients), and European LeukemiaNet (ELN) high-risk score (ET and PV patients).3,16 -19
Statistics
Significance was defined as P < .05. Baseline characteristics were reported as median, range, and 95% confidence interval and compared using Student 2-tailed t test, χ2 analysis, and Fisher’s exact test, as appropriate. Hemorrhage-free survival, overall survival, and thrombosis-free survival curves were obtained using Kaplan-Meier methods. Risk factors for thrombosis, bleeding, and evolution were investigated in univariate analysis; all variables with P < .1 were included in a logistic regression model. Data analysis was performed using R version 3.6.2 (The R Foundation for Statistical Computing, Vienna, Austria) and SPSS version 23.0 (IBM Corp., Armonk, NY).20-22
Results
General information
Altogether, 444 patients from 15 countries (supplemental Figure 1), median follow-up 9.7 years (0-47.8), were recruited. Most were diagnosed between 1990 and 2019 (80%). Age at diagnosis between 20 and 25 years (n = 239, 53.8%) was most frequent, but 51 (11.5%) patients were younger than 15 years at presentation (supplemental Figure 2). MPN subtype was ET in 318 (71.6%), PV in 81 (18.2%), PMF in 21 (4.7%), PreMF in 11 (2.5%), and MPN-u in 13 (2.9%) patients; PMF, PreMF, and MPN-u were grouped as “other MPNs” for analysis.
Clinical and biological data at diagnosis
Clinical characteristics.
The median age at diagnosis of MPN was ∼20 years old, and the proportion of women was higher in the ET and “other MPNs” subgroups compared with PV (>75% vs 48.1%) (Table 1). A familial history of hematological disease was identified in 25 of 409 patients (6.1%).
Characteristics of the population at diagnosis
| Parameters | Cohort | ET | PV | Other MPNs | ||||
|---|---|---|---|---|---|---|---|---|
| Nb/median | % or range | Nb/median | % or range | Nb/median | % or range | Nb/median | % or range | |
| Numbers | 444 | 318 | 71.6 | 81 | 18.2 | 45 | 10.2 | |
| Female | 321 | 72.3 | 248 | 78 | 39 | 48.1 | 34 | 75.6 |
| Age | 20.4 | 2-25 | 20.6 | 2-25 | 19.7 | 2.3-25 | 20.1 | 5.3-24.4 |
| Symptoms | ||||||||
| Plethoric face | 15 | 3.9 | 2 | 0.7 | 13 | 21.3 | 0 | 0 |
| Aquagenic pruritus | 22 | 5.6 | 9 | 3.2 | 13 | 19.7 | 0 | 0 |
| Hyperviscosity | 127 | 34.5 | 91 | 34.2 | 27 | 42.2 | 9 | 23.7 |
| Microvascular symptoms | 43 | 10.8 | 33 | 11.6 | 4 | 5.8 | 6 | 14 |
| Constit symptoms | 24 | 6.2 | 11 | 3.9 | 6 | 9.2 | 7 | 16.7 |
| Palpable splenomegaly | 80 | 20.3 | 39 | 13.6 | 25 | 39.1 | 16 | 37.2 |
| Fatigue | 73 | 19.8 | 44 | 16.5 | 20 | 34.5 | 9 | 20.9 |
| Cardiovascular risk factors | 56 | 12.9 | 38 | 12.2 | 12 | 15.4 | 6 | 13.6 |
| Biology | ||||||||
| Leukocytes (×109/L) | 9 | 2.8-28.1 | 8.9 | 3-28.1 | 9.8 | 2.8-25 | 10.3 | 3-20.9 |
| Hemoglobin (g/L) | 140 | 65-246 | 139 | 99-171 | 174.5 | 134-246 | 132 | 65-168 |
| Platelets (×109/L) | 863 | 99-3290 | 915 | 181-3290 | 601 | 160-1170 | 775 | 99-2036 |
| Mutations | ||||||||
| JAK2V617F | 249 | 56 | 154 | 48.5 | 70 | 86.4 | 25 | 55.6 |
| JAK2 ex 12 | 5 | 1.13 | 0 | 0 | 5 | 6.2 | 0 | 0 |
| JAK2 allele burden | 22 | 1.75-86 | 15.5 | 1.75-86 | 29 | 3.7-77 | 31.8 | 11.8-49 |
| CALR | 59 | 13.3 | 46 | 14.5 | 0 | 0 | 13 | 28.9 |
| MPL | 3 | 0.7 | 3 | 0.9 | 0 | 0 | 0 | 0 |
| Triple negativity | 89 | 20 | 86 | 27 | 0 | 0 | 3 | 6.7 |
| Incomplete or unknown | 39 | 8.8 | 29 | 9.1 | 6 | 7.4 | 4 | 0 |
| Parameters | Cohort | ET | PV | Other MPNs | ||||
|---|---|---|---|---|---|---|---|---|
| Nb/median | % or range | Nb/median | % or range | Nb/median | % or range | Nb/median | % or range | |
| Numbers | 444 | 318 | 71.6 | 81 | 18.2 | 45 | 10.2 | |
| Female | 321 | 72.3 | 248 | 78 | 39 | 48.1 | 34 | 75.6 |
| Age | 20.4 | 2-25 | 20.6 | 2-25 | 19.7 | 2.3-25 | 20.1 | 5.3-24.4 |
| Symptoms | ||||||||
| Plethoric face | 15 | 3.9 | 2 | 0.7 | 13 | 21.3 | 0 | 0 |
| Aquagenic pruritus | 22 | 5.6 | 9 | 3.2 | 13 | 19.7 | 0 | 0 |
| Hyperviscosity | 127 | 34.5 | 91 | 34.2 | 27 | 42.2 | 9 | 23.7 |
| Microvascular symptoms | 43 | 10.8 | 33 | 11.6 | 4 | 5.8 | 6 | 14 |
| Constit symptoms | 24 | 6.2 | 11 | 3.9 | 6 | 9.2 | 7 | 16.7 |
| Palpable splenomegaly | 80 | 20.3 | 39 | 13.6 | 25 | 39.1 | 16 | 37.2 |
| Fatigue | 73 | 19.8 | 44 | 16.5 | 20 | 34.5 | 9 | 20.9 |
| Cardiovascular risk factors | 56 | 12.9 | 38 | 12.2 | 12 | 15.4 | 6 | 13.6 |
| Biology | ||||||||
| Leukocytes (×109/L) | 9 | 2.8-28.1 | 8.9 | 3-28.1 | 9.8 | 2.8-25 | 10.3 | 3-20.9 |
| Hemoglobin (g/L) | 140 | 65-246 | 139 | 99-171 | 174.5 | 134-246 | 132 | 65-168 |
| Platelets (×109/L) | 863 | 99-3290 | 915 | 181-3290 | 601 | 160-1170 | 775 | 99-2036 |
| Mutations | ||||||||
| JAK2V617F | 249 | 56 | 154 | 48.5 | 70 | 86.4 | 25 | 55.6 |
| JAK2 ex 12 | 5 | 1.13 | 0 | 0 | 5 | 6.2 | 0 | 0 |
| JAK2 allele burden | 22 | 1.75-86 | 15.5 | 1.75-86 | 29 | 3.7-77 | 31.8 | 11.8-49 |
| CALR | 59 | 13.3 | 46 | 14.5 | 0 | 0 | 13 | 28.9 |
| MPL | 3 | 0.7 | 3 | 0.9 | 0 | 0 | 0 | 0 |
| Triple negativity | 89 | 20 | 86 | 27 | 0 | 0 | 3 | 6.7 |
| Incomplete or unknown | 39 | 8.8 | 29 | 9.1 | 6 | 7.4 | 4 | 0 |
Constit, constitutional; ex, exon; MPL, myeloproliferative leukemia; Nb, number.
Only 149 patients (33.3%) were asymptomatic. The most frequent symptoms were hyperviscosity (34.5%) and fatigue (19.8%). Microvascular or constitutional symptoms were less frequent (10.8% and 6.2%, respectively). Hyperviscosity symptoms included headache (73%), vertigo/dizziness (18%), and blurred vision (11.8%). Symptom profiles were different between diseases. PV patients were the most symptomatic, with hyperviscosity (42.2%), fatigue (34.5%), plethoric face (21.3%), and aquagenic pruritus (19.7%), whereas patients with other MPNs mostly expressed constitutional symptoms (16.7%).
Palpable splenomegaly was reported in 20% of patients (mostly in PV and “other MPNs”: 39.1% and 37.2%, respectively). Ultrasound scan data were not requested. Cardiovascular risk factors were found in 56 of 434 cases (12.9%), with the most frequent being smoking (40 patients, 91.4%), hypertension (8% to 14.3%), and obesity (5% to 8.9%).
Mutational profile.
Driver mutation status was available in 405 patients (91.2%). The mutational landscape is shown in Figure 1A. The JAK2V617F mutation was predominant in all MPN. In PV, 6.2% of patients had a JAK2 exon 12 mutation. Concerning the ET population, 48.5% of patients had JAK2V617F, 14.5% CALR, and 0.9% MPL mutations, whereas 27% were triple negative. We observed a clear difference across age groups, with most triple-negative cases observed in children and a majority of JAK2V617F cases in adolescents/young adults (Figure 1B). In the “other” group, 55.6% of cases were JAK2V617F and 28.9% CALR, and only 6.7% were triple negatives. The median JAK2V617F mutant allele burden was 22% in the whole cohort and higher in :other MPN” (31.8%) and PV (29%) compared with ET patients (15.5%) (Figure 1C).
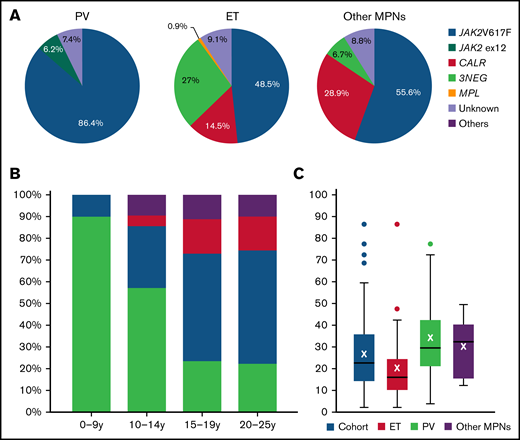
Driver mutation status in the population of children, adolescents, and young adults. (A) Driver mutation status depending on MPN subtype in the global population. (B) Driver mutations observed in the ET population depending on age. (C) JAK2V617F allele burden at diagnosis of MPN. 3NEG, triple negative; CALR, calreticulin; JAK2, just another kinase 2; Others, MPL or unknown.
Driver mutation status in the population of children, adolescents, and young adults. (A) Driver mutation status depending on MPN subtype in the global population. (B) Driver mutations observed in the ET population depending on age. (C) JAK2V617F allele burden at diagnosis of MPN. 3NEG, triple negative; CALR, calreticulin; JAK2, just another kinase 2; Others, MPL or unknown.
Maternity
Overall, 119 women (41.2%) experienced 214 pregnancies, with 37.5%, 42.9%, and 36.7% in ET, PV, and “other MPNs,” respectively. The live birth rate of 78% was similar between disease groups at 80.1%, 68.8%, and 76.2% in ET, PV, and “other MPNs,” respectively.
Therapeutic strategy
During follow-up, 301 (67.8%) received at least 1 cytoreductive drug, and 333 (75%) received antiplatelet or anticoagulant drug. Altogether, 47 patients received no drug (10.6%), 64 patients only a cytoreductive drug (14.4%) and 96 patients only an antithrombotic drug (21.6%). Phlebotomies were prescribed in 73 PV patients.
Antithrombotic drugs were most prescribed to 243 ET patients (76.4%) and 70 PV patients (86.4%) compared with 20 “other MPNs” patients (44.4%) (P = .00002). Cytoreductive drugs were prescribed to 217 ET (68.2%), 58 PV (71.6%), and 26 “other MPNs” patients (57.8%). Cytoreductive and antithrombotic drugs were given to 170 ET patients (53.5%), 52 PV patients (64.2%), and 15 “other MPNs” patients (33.3%) (P = .0002).
Rationale for starting cytoreduction was available for 156 patients (51.8%): platelet count > 1000 × 109/L (55%), pregnancy (19%), phenotypic evolution (15%), and symptoms (11%). According to current ELN criteria, only 95 patients (21.4%) were theoretically eligible for cytoreduction: 49 because of a platelet count > 1500 × 109/L, 49 for a history of thrombosis, and 3 patients for both reasons. Over the follow-up period, 29% had 1 cytoreductive agent, 23% 2, and 16% ≥3. As first-line treatment, hydroxycarbamide was the most prescribed (52.2%), whereas interferon was the most drug used in second (57.2%) and third lines (43.5%) (Figure 2). Ruxolitinib was prescribed in 11.6% of the cases, only as third line.

Distribution of cytoreductive drug used as first, second, or third line. SCT, stem cell transplantation.
Distribution of cytoreductive drug used as first, second, or third line. SCT, stem cell transplantation.
An allogeneic stem cell transplant was performed in 7 patients: 5 for progressive MF and 2 after transformation to AP or AML.
Antiplatelet therapies were prescribed in 277 patients (83.2%): low dose aspirin (LDA) in 267 (80.2%), clopidogrel in 7 (2.1%), and both in 3 (0.9%). Vitamin K antagonists (VKAs) were given to 44 patients (13.2%) (6 VKA and LDA); direct oral anticoagulants (DOAC) and low-molecular-weight heparins were each prescribed in 6 cases (1.8%), and 2 patients per group received DOAC and LDA.
Thrombosis
Forty-nine patients (11.1%) had a history of thrombosis: 27 ET (8.5% of ET cohort), 17 PV (21.5% of PV cohort), and 5 “other MPNs” (11.4%, (P = .007) (Table 2).
Thrombosis, hemorrhage, and phenotypic evolution: history and risk during the follow-up in the population
| Cohort | ET | PV | Other MPNs | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Parameters | Nb or median | % or range | Nb or median | % or range | Nb or median | % or range | Nb or median | % or range | P | |
| History of thromboses | 49 | 11.1 | 27 | 8.5 | 17 | 21.5 | 5 | 11.4 | .007 | |
| New thromboses | 49 | 11.1 | 32 | 10.1 | 13 | 16.3 | 4 | 9.1 | .27 | |
| Median time | 5 | 0.1-28.5 | 4.6 | 0.2-28.5 | 4.5 | 0.1-19.4 | 9.6 | 4.1-18.2 | ||
| Incidence (%) | At 1 y | 6 | 1.36 | 4 | 1.26 | 2 | 2.5 | 0 | — | |
| At 5 y | 25 | 5.67 | 17 | 5.36 | 7 | 8.75 | 1 | 2.27 | ||
| At 10 y | 35 | 7.94 | 25 | 7.89 | 8 | 10 | 2 | 4.55 | ||
| Incidence (% pt/y) | 1.16 | 1.14 | 1.31 | 0.94 | ||||||
| Recurrence | 17 | 34.7 | 11 | 34.4 | 4 | 30.8 | 2 | 50 | .8 | |
| History of hemorrhages | 25 | 5.7 | 19 | 6 | 4 | 5.1 | 2 | 4.5 | 1 | |
| New hemorrhages | 44 | 9.9 | 27 | 8.5 | 11 | 13.8 | 6 | 13.6 | .23 | |
| Median time | 4.65 | 0.06-35.1 | 3.25 | 0.06-20.98 | 12.5 | 1.64-35.1 | 8.4 | 1-19.21 | ||
| Incidence (%) | At 1 y | 7 | 1.6 | 6 | 1.93 | 0 | — | 1 | 2.27 | .44 |
| At 5 y | 19 | 4.37 | 14 | 4.5 | 3 | 3.8 | 3 | 6.8 | .7 | |
| At 10 y | 24 | 5.52 | 18 | 5.79 | 3 | 3.8 | 3 | 6.8 | .78 | |
| Incidence (% pt/y) | 1.04 | 0.94 | 1.17 | 1.4 | ||||||
| Evolutions | Total | 48 | 10.9 | 33 | 10.4 | 8 | 10 | 7 | 15.6 | .5 |
| PV | 12 | 2.7 | 9 | 2.8 | — | — | 3 | 6.7 | .17 | |
| MF | 33 | 7.5 | 23 | 7.3 | 7 | 8.8 | 3 | 6.7 | .91 | |
| AP/AML | 3 | 0.7 | 1 | 0.3 | 1 | 1.2 | 1 | 2.2 | .19 | |
| Incidence PV (%) | At 1 y | 0 | — | 0 | — | NA | NA | 0 | — | |
| At 5 y | 3 | 0.68 | 1 | 0.31 | NA | NA | 2 | 4.44 | .04 | |
| At 10 y | 6 | 1.36 | 4 | 1.26 | NA | NA | 2 | 4.44 | .16 | |
| Incidence MF (%) | At 1 y | 0 | — | 0 | — | 0 | — | 0 | — | |
| At 5 y | 11 | 2.49 | 6 | 1.89 | 3 | 3.75 | 2 | 4.44 | .29 | |
| At 10 y | 18 | 4.07 | 12 | 3.79 | 4 | 5 | 2 | 4.44 | .73 | |
| Incidence (% pt/y) | Total | 1.13 | 1.14 | 0.84 | 1.63 | |||||
| PV | 0.28 | 0.31 | 0 | 0.69 | ||||||
| MF | 0.77 | 0.8 | 0.73 | 0.69 | ||||||
| AP/AML | 0.07 | 0.03 | 0.03 | 0.23 | ||||||
| Cohort | ET | PV | Other MPNs | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Parameters | Nb or median | % or range | Nb or median | % or range | Nb or median | % or range | Nb or median | % or range | P | |
| History of thromboses | 49 | 11.1 | 27 | 8.5 | 17 | 21.5 | 5 | 11.4 | .007 | |
| New thromboses | 49 | 11.1 | 32 | 10.1 | 13 | 16.3 | 4 | 9.1 | .27 | |
| Median time | 5 | 0.1-28.5 | 4.6 | 0.2-28.5 | 4.5 | 0.1-19.4 | 9.6 | 4.1-18.2 | ||
| Incidence (%) | At 1 y | 6 | 1.36 | 4 | 1.26 | 2 | 2.5 | 0 | — | |
| At 5 y | 25 | 5.67 | 17 | 5.36 | 7 | 8.75 | 1 | 2.27 | ||
| At 10 y | 35 | 7.94 | 25 | 7.89 | 8 | 10 | 2 | 4.55 | ||
| Incidence (% pt/y) | 1.16 | 1.14 | 1.31 | 0.94 | ||||||
| Recurrence | 17 | 34.7 | 11 | 34.4 | 4 | 30.8 | 2 | 50 | .8 | |
| History of hemorrhages | 25 | 5.7 | 19 | 6 | 4 | 5.1 | 2 | 4.5 | 1 | |
| New hemorrhages | 44 | 9.9 | 27 | 8.5 | 11 | 13.8 | 6 | 13.6 | .23 | |
| Median time | 4.65 | 0.06-35.1 | 3.25 | 0.06-20.98 | 12.5 | 1.64-35.1 | 8.4 | 1-19.21 | ||
| Incidence (%) | At 1 y | 7 | 1.6 | 6 | 1.93 | 0 | — | 1 | 2.27 | .44 |
| At 5 y | 19 | 4.37 | 14 | 4.5 | 3 | 3.8 | 3 | 6.8 | .7 | |
| At 10 y | 24 | 5.52 | 18 | 5.79 | 3 | 3.8 | 3 | 6.8 | .78 | |
| Incidence (% pt/y) | 1.04 | 0.94 | 1.17 | 1.4 | ||||||
| Evolutions | Total | 48 | 10.9 | 33 | 10.4 | 8 | 10 | 7 | 15.6 | .5 |
| PV | 12 | 2.7 | 9 | 2.8 | — | — | 3 | 6.7 | .17 | |
| MF | 33 | 7.5 | 23 | 7.3 | 7 | 8.8 | 3 | 6.7 | .91 | |
| AP/AML | 3 | 0.7 | 1 | 0.3 | 1 | 1.2 | 1 | 2.2 | .19 | |
| Incidence PV (%) | At 1 y | 0 | — | 0 | — | NA | NA | 0 | — | |
| At 5 y | 3 | 0.68 | 1 | 0.31 | NA | NA | 2 | 4.44 | .04 | |
| At 10 y | 6 | 1.36 | 4 | 1.26 | NA | NA | 2 | 4.44 | .16 | |
| Incidence MF (%) | At 1 y | 0 | — | 0 | — | 0 | — | 0 | — | |
| At 5 y | 11 | 2.49 | 6 | 1.89 | 3 | 3.75 | 2 | 4.44 | .29 | |
| At 10 y | 18 | 4.07 | 12 | 3.79 | 4 | 5 | 2 | 4.44 | .73 | |
| Incidence (% pt/y) | Total | 1.13 | 1.14 | 0.84 | 1.63 | |||||
| PV | 0.28 | 0.31 | 0 | 0.69 | ||||||
| MF | 0.77 | 0.8 | 0.73 | 0.69 | ||||||
| AP/AML | 0.07 | 0.03 | 0.03 | 0.23 | ||||||
Nb, number; NA, not applicable.
Postdiagnosis, 49 patients (11.1%) suffered from thrombosis, mostly in PV (n = 13, 16.3%) but not different between groups (10.1% in ET and 9.1% in “other MPNs”). The global incidence of thrombosis was 1.16% patient per year (pt/y) (1.31% in PV, 1.14% in ET, and 0.94% in “other MPNs”). The 5-year incidence of thrombosis was 5.67% in the whole cohort, with 5.36%, 8.75%, and 2.27% in ET, PV, and “other MPNs,” respectively. The median time from diagnosis to first thrombotic event was 5 years, and the median time between the first new and the second new event was 4.4 years. One-third experienced recurrent thrombotic events without differences between groups. Venous events were more frequent (n = 82; 71.3%) than arterial (n = 29; 25.2%) (supplemental Table 1). Overall, perihepatic vein thromboses were most common (n = 39; 47.6% of venous events) compared with deep vein thrombosis/pulmonary embolism (n = 18; 22%) or cerebral vein thromboses (n = 16; 19.5%). Prevalence of Budd-Chiari syndrome and cerebral vein thrombosis before/at diagnosis was high (36.1% and 33.3%, respectively) compared with portal/splanchnic vein thromboses and deep vein thrombosis/pulmonary embolism after diagnosis (23% and 18.5%, respectively; P = .03). No such differences were observed for arterial events. Interestingly, both splanchnic vein thromboses (26/41 cases, 63.4%) and total of venous events (57/72 cases, 79.2%) were most frequent in females.
Concerning thrombosis-free survival, there was no difference between MPN subtypes; however, JAK2+ ET patients had shorter median time to thrombosis (28.5 years) compared with other ET patients (Figure 3A-B). Across the cohort, JAK2V617F mutation (odds ratio [OR], 3.31 [1.61;6.82],P = .001), hyperviscosity symptoms (OR, 2.70 [1.27;5.75], P = .015), and thrombosis history (OR, 2.38 [1.10;5.15], P = .047) were significant predictive factors, but that was not true for constitutional symptoms (OR, 2.62 [0.92;7.48], P = .074) or cardiovascular risk factors (OR, 2.07 [0.96;1.44], P = .065). In the logistic regression model, JAK2V617F mutation (OR, 3.496 [1.26;9.66], P = .016) and hyperviscosity symptoms (OR, 2.37 [1.04;5.41], P = .04) remained significant. For ET, JAK2V617F (OR, 3.05 [1.36;6.82], P = .005) and hyperviscosity symptoms (OR, 3.17 [1.25;8.07], P = .015) were significant, with a trend toward significance for constitutional symptoms (OR, 4.01 [0.99;16.17], P = .071). In logistic regression model, JAK2V617F (OR, 3.40 [1.15;10.09], P = .027) and hyperviscosity symptoms (OR, 2.98 [1.12;7.90], P = .028) remained significant. For PV patients, cardiovascular risk factors (OR, 3.63 [0.89;14.84], P = .082) and leukocytes > 11 g/L (OR, 6.36 [0.69;58.5], P = .096) trended toward significance in univariate analysis but were not confirmed in logistic regression. All informations are summarized in Table 3. Analyses were not performed in the “other MPNs” group due to low number of events.
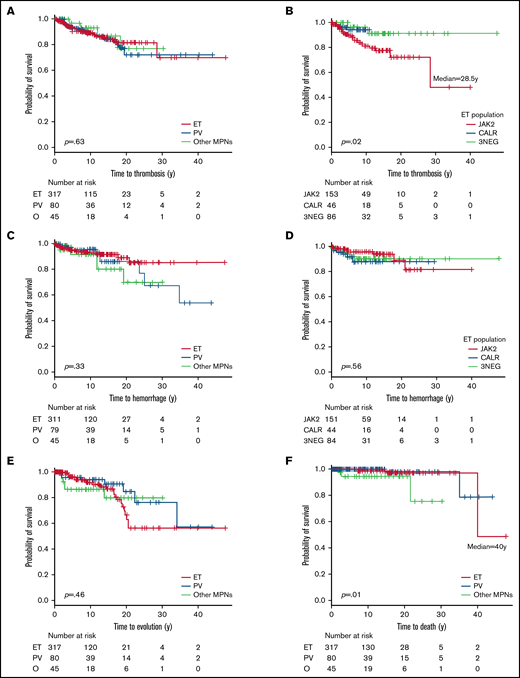
Kaplan-Meier curves of complications during follow-up of the global cohort. Thrombosis-free survival curves in all MPN (A) and in the ET population (B). Hemorrhage-free survival curves in all MPN (C) and in the ET population (D). Evolution-free survival curves (E) and overall survival curves (F). 3NEG, triple negative; NR, not reached; O, other MPNs.
Kaplan-Meier curves of complications during follow-up of the global cohort. Thrombosis-free survival curves in all MPN (A) and in the ET population (B). Hemorrhage-free survival curves in all MPN (C) and in the ET population (D). Evolution-free survival curves (E) and overall survival curves (F). 3NEG, triple negative; NR, not reached; O, other MPNs.
Thrombosis, bleeding, and evolution risk factors: multivariate analyses
| Risk factors for thrombosis | Whole cohort | ET | PV | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Multivariate analysis | OR | 95% CI | P | OR | 95% CI | P | OR | 95% CI | P |
| JAK2 vs other mutations | 3.49 | 1.26-9.66 | .016 | 3.40 | 1.15-10.09 | .027 | NA | ||
| Hyperviscosity vs no | 2.37 | 1.04-5.41 | .040 | 2.98 | 1.12-7.90 | .028 | NA | ||
| Constitutional symptoms vs no | 2.11 | 0.55-8.17 | .279 | 3.43 | 0.60-19.79 | .167 | NA | ||
| Cardiovascular RF vs no | 1.60 | 0.50-5.09 | .430 | NA | 3.35 | 0.25-45.37 | .363 | ||
| Thrombosis history vs no | 0.93 | 0.26-3.40 | .916 | NA | NA | ||||
| Leukocytes > 11 g/L vs no | NA | NA | 6.30 | 0.67-59.07 | .107 | ||||
| Risk factors for thrombosis | Whole cohort | ET | PV | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Multivariate analysis | OR | 95% CI | P | OR | 95% CI | P | OR | 95% CI | P |
| JAK2 vs other mutations | 3.49 | 1.26-9.66 | .016 | 3.40 | 1.15-10.09 | .027 | NA | ||
| Hyperviscosity vs no | 2.37 | 1.04-5.41 | .040 | 2.98 | 1.12-7.90 | .028 | NA | ||
| Constitutional symptoms vs no | 2.11 | 0.55-8.17 | .279 | 3.43 | 0.60-19.79 | .167 | NA | ||
| Cardiovascular RF vs no | 1.60 | 0.50-5.09 | .430 | NA | 3.35 | 0.25-45.37 | .363 | ||
| Thrombosis history vs no | 0.93 | 0.26-3.40 | .916 | NA | NA | ||||
| Leukocytes > 11 g/L vs no | NA | NA | 6.30 | 0.67-59.07 | .107 | ||||
| Risk factors for bleeding | Whole cohort | ET | PV | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Multivariate analysis | OR | 95% CI | P | OR | 95% CI | P | OR | 95%CI | P |
| Hyperviscosity vs no | 3.06 | 1.24-7.56 | .015 | 7.40 | 2.10-26.10 | .002 | NA | ||
| Palpable splenomegaly vs no | 2.82 | 1.33-7.04 | .026 | NA | NA | ||||
| Cardiovascular RF vs no | 1.74 | 0.54-5.62 | .355 | NA | NA | ||||
| Bleeding history vs no | 10.93 | 3.23-37.07 | .000 | 48.79 | 9.30-256.05 | .000 | NA | ||
| Platelets > 1500 g/L vs no | 2.92 | 1.09-7.87 | .034 | 4.97 | 1.37-18.05 | .002 | NA | ||
| Age < 20 y vs no | NA | 0.09 | 0.02-0.43 | .002 | NA | ||||
| Risk factors for bleeding | Whole cohort | ET | PV | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Multivariate analysis | OR | 95% CI | P | OR | 95% CI | P | OR | 95%CI | P |
| Hyperviscosity vs no | 3.06 | 1.24-7.56 | .015 | 7.40 | 2.10-26.10 | .002 | NA | ||
| Palpable splenomegaly vs no | 2.82 | 1.33-7.04 | .026 | NA | NA | ||||
| Cardiovascular RF vs no | 1.74 | 0.54-5.62 | .355 | NA | NA | ||||
| Bleeding history vs no | 10.93 | 3.23-37.07 | .000 | 48.79 | 9.30-256.05 | .000 | NA | ||
| Platelets > 1500 g/L vs no | 2.92 | 1.09-7.87 | .034 | 4.97 | 1.37-18.05 | .002 | NA | ||
| Age < 20 y vs no | NA | 0.09 | 0.02-0.43 | .002 | NA | ||||
| Risk factors for evolution | Whole cohort | ET | PV | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Multivariate analysis | OR | 95% CI | P | OR | 95%CI | P | OR | 95% CI | P |
| Palpable splenomegaly vs no | NA | 8.42 | 3.58-19.77 | .000 | NA | ||||
| Cardiovascular RF vs no | NA | 3.98 | 1.48-10.68 | .006 | NA | ||||
| Thrombosis history vs no | NA | NA | 5.88 | 1.15-30.09 | .033 | ||||
| Age < 20 y vs no | NA | NA | 9.63 | 1.04-88.79 | .046 | ||||
| Risk factors for evolution | Whole cohort | ET | PV | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Multivariate analysis | OR | 95% CI | P | OR | 95%CI | P | OR | 95% CI | P |
| Palpable splenomegaly vs no | NA | 8.42 | 3.58-19.77 | .000 | NA | ||||
| Cardiovascular RF vs no | NA | 3.98 | 1.48-10.68 | .006 | NA | ||||
| Thrombosis history vs no | NA | NA | 5.88 | 1.15-30.09 | .033 | ||||
| Age < 20 y vs no | NA | NA | 9.63 | 1.04-88.79 | .046 | ||||
RF, risk factors; NA, not applicable.
Hemorrhage
Twenty-five patients (5.7%) had a history of hemorrhage, most commonly ET patients (n = 19; 6%) (Table 2). New hemorrhagic events were observed in 44 patients (9.9%): 8.5% of ET, 13.8% of PV, and 13.6% of “other MPNs.” Overall hemorrhage incidence was 1.04% pt/y, between 0.94% in ET and 1.4% in “other MPNs.” The 5-year incidence was 4.37% in the whole cohort, with 4.5%, 3.8%, and 6.8% in ET, PV, and “other MPNs,” respectively. The median time from diagnosis to first hemorrhagic event was 4.7 years. In terms of hemorrhage-free survival, no difference was seen between MPN subgroups and between mutated vs nonmutated ET (Figure 3C-D).
Regarding risk factors for hemorrhage, in univariate analysis in the whole cohort, hyperviscosity symptoms (OR, 2.17 [1.07;4.37], P = .039), splenomegaly (OR, 3.05 [1.50;6.20], P = .004), bleeding history (OR, 11.05 [4.66;26.20], P = .000), and platelet count > 1500 g/L (OR, 2.61 [1.14;5.98], P = .031) were associated with significantly increased risk of bleeding. In addition, cardiovascular risk factors (OR, 2.20 [1.02;4.75], P = .055) had a trend toward significance. In the logistical regression model, hyperviscosity symptoms (OR, 3.06 [1.24;7.56], P = .015), splenomegaly (OR, 2.82 [1.33;7.04], 0.026), bleeding history (OR, 10.93 [3.23;37.07], P = .000) and platelet count > 1500 g/L (OR, 2.92 [1.09;7.87], P = .034) remained significant. For ET patients, hyperviscosity symptoms (OR, 2.75 [1.16;6.55], P = .022) and bleeding history (OR, 14 [5.05;38.78], P < .001) were significant; platelet count > 1500 g/L (OR, 2.64 [1.02;6.85], P = .064) and age < 20 years (OR, 0.43 [0.17;1.04], P = .067) had a trend toward significance. In the logistic regression model, all variables were significant, particularly bleeding history (OR, 48.8 [9.30;256.05], P = .000). For PV patient, no significant predictive factors were identified for bleeding using either model (Table 3).
Phenotypic evolution
We observed 48 phenotypic evolutions (10.9%): 10% in ET and PV and 15.6% in “other MPNs” (Table 2). Global incidence was 1.13% pt/y, from 0.84 in PV to 1.63 in “other MPNs.” The most common transformation was evolution to SMF (n = 33%-7.5%), observed in 8.8% of PV, 7.3% of ET, and 6.7% of “other MPNs.” The 5-year incidence was 2.49% in the whole cohort (1.89% in ET and 3.75% in PV). Transformation to PV occurred in 12 patients (2.7%), mostly from ET (n = 9%-2.8%), and all were JAK2V617F+. AP and AML were observed in 3 patients (0.7%). Evolution-free survival curves are shown in Figure 3E. Median time to evolution was not reached.
Regarding risk factors for evolution (Table 3), in univariate analysis, only palpable splenomegaly (OR, 3.45 [1.80;6.63], P = .000) was significant in the whole cohort. In ET patients, palpable splenomegaly (OR, 7.61 [3.36;17.26], P < .001) and cardiovascular risk factors (OR, 2.66 [1.10;6.41], P = .043) were associated with phenotypic evolution and remained significant in the logistic regression model. In PV patients, thrombosis history (OR, 4.46 [0.99;20.21], P = .061) and age < 20 years (OR, 7.40 [0.87;63.25], P = .059) had a trend toward significance becoming significant in logistic regression model.
Deaths
Eight deaths (1.8%) were recorded, incidence ranging from 0.9% in ET to 6.7% in “other MPNs” (P = .022) (Figure 3F). Three patients died after allogeneic stem cell transplantation. Causes of deaths were bleeding (n = 2), leukemia, solid cancer (high-grade astrocytoma in patient aged 60 years), hepatic failure, graft-versus-host disease, and cytomegalovirus pneumonia, not described (n = 1 each).
Performance of current risk scores
Performance of conventional risk scores for survival and thrombosis was assessed. ET patients were classified according to ELN, IPSET-T, and IPSET-NT scores. IPSET-T and IPSET-NT scores were the only scores able to differentiate patients in terms of thrombotic risk. The IPSET-T score applied to the ET population shows a thrombotic risk of 23.7%, 13%, and 4.3% in high, intermediate, and low groups (P = .0009), respectively. The IPSET-NT score shows a thrombotic risk of 18.5%, 14.5%, and 4.7% in high, low, and very low groups (P = .004), respectively. ELN score was not discriminatory (supplemental Tables 2 and 3).
Similarly, for Kaplan-Meier estimation according to the IPSET-T and IPSET-NT scores, only high-risk patients had an available median-free survival of 28.5 years (for both). Differences between groups of patients were highly significant (P = .001 and P = .004, respectively); patients were also differentiated by ELN score (P = .056) (Figure 4A-C).
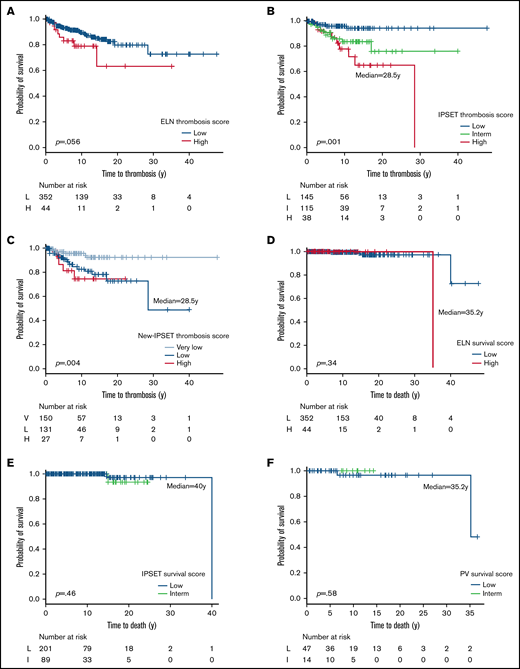
Kaplan-Meier curves for thrombosis and survival scores applied to children, adolescents, and young adults’ population. Survival and thrombosis scores: (A) ELN score and thrombosis, (B) IPSET-T score, (C) IPSET-NT score, (D) ELN score and survival, (E) IPSET survival score, and (F) PV survival score. H, high risk; I, intermediate risk; L, low risk.
Kaplan-Meier curves for thrombosis and survival scores applied to children, adolescents, and young adults’ population. Survival and thrombosis scores: (A) ELN score and thrombosis, (B) IPSET-T score, (C) IPSET-NT score, (D) ELN score and survival, (E) IPSET survival score, and (F) PV survival score. H, high risk; I, intermediate risk; L, low risk.
Concerning survival, no score was able to differentiate PV patients (supplemental Table 3; Figure 4D-F).
Discussion
We studied a large cohort of 444 patients from 15 countries diagnosed with MPN before the age of 25. The median age was 20.4 years, reflecting most cases were diagnosed in young adults, and 11.5% were diagnosed before 10 years old.
We confirm that ET is the most frequent MPN in young persons (71.6% in this cohort; 84% in our previous review).9 Importantly, patients with familial thrombocytosis were excluded from this cohort. We observed some cases of PreMF, which is rare compared with adult populations,23,24 underscoring the importance of histopathology to classify MPN.4-20 Here, 68.7% of patients had informative bone marrow biopsy (including for PV diagnosis), less than expected if current World Health Organization guidelines were employed. Molecular analyses were available in 92.1% of patients. In PV, a JAK2 mutation was found in 93% of patients, including 6.7% JAK2 exon 12 mutation, a higher proportion than observed in adults (<3%).25,26 In ET and PMF/PreMF/MPN-u cases, proportions of driver mutations and allelic burdens seem to be in accordance with previous publications.27-29 Interestingly, the distribution of driver mutations changes dramatically with the age of patients at diagnosis, from 90% lacking any driver mutations in the youngest children to 22.2% for younger adults (Figure 1). The high rate of triple-negative ET cases in women is in accordance with previous literature.28 Future studies to centrally analyze the available bone marrow biopsies and archival DNA to assess the incidence and impact of additional mutations detected by next generation sequencing analyses are planned.30
Our data provide interesting perspectives on how these MPNs in young patients are perceived by physicians (in terms of risk and prevention) and how this population is managed. There are no specific recommendations in national or international guidelines about how to treat such young patients with MPN. Importantly, we demonstrate that standard prognostic scores do not perform well in this cohort. As an example, 67.8% received at least 1 cytoreductive drug when only 21.4% of them should have received such therapy according to current ELN recommendations (patients with a history of thrombosis and/or platelet count > 1500 × 109/L). The reported reasons to prescribe cytoreductive therapies in these young patients were platelet count > 1000 × 109/L in 55% and symptoms in 11%. Proportions of hydroxycarbamide (52.2%), anagrelide (22.9%), and interferon (22.6%) prescriptions also suggest that physicians did not apply ELN recommendations where interferon is the recommended first line therapy in patients aged <60 years old because of its lack of leukemogenicity and its low teratogenicity.3,31 However, no current guidelines exist for MPN in children or adolescents. These differences in thresholds for therapy and choice of drug likely reflect historical shifts in guidelines and availability of therapy. Interestingly, 29% of the patients stopped interferon due to intolerance, compared with 38.5% in the literature.32 Hydroxycarbamide was the most prescribed drug despite the age of these patients.33-36 Here, only 2 evolutions to AML were observed, and only 1 of these patients was previously treated by hydroxycarbamide, though due to our median follow-up, this observation should not be overinterpreted. Low-dose aspirin was the most frequently prescribed antithrombotic agent (80.2%), and importantly, no case of Reye syndrome was reported.37 This number seems higher than expected in this very young population. There is no recommendation (indeed no trial either) about the use of antiplatelets or anticoagulant drugs in these populations. This study was not performed to assess efficacy of such drugs in terms of complete or partial hematological responses and/or reduction of complication rates. In chronic myeloid leukemia, it has been observed that adolescents and young adults have lower rates of complete cytogenetic and molecular responses.38,39 One explanation could be the lower observance rate in this population.40
The risk of vascular complications is often perceived as lower in younger MPN patients compared with the adult MPN population. In our literature review of ET or PV patients diagnosed before 20 years, reported incidences of thrombosis, hemorrhage, and transformation were 4.7%, 4.7%, and 1.9%, respectively.9 In the current “real-world” cohort, considering only ET and PV cases, these incidences were 13.8%, 9.5%, and 7.5% (Table 2), respectively, so 3, 2, and 4 times higher. These numbers are therefore completely different than those previously reported in smaller cohorts and closer to those observed in adults and older patients.41
In terms of thrombosis, we observed that 11% of patients had a history of thrombotic events, particularly in PV (21.5%), which is somewhat surprising for this very young population. Interestingly, most of the events were venous (71.3%) and equally observed before and after diagnosis, whereas this incidence is around 40% to 50% in adults MPN.35,42 Perihepatic thromboses were the most frequent (47.6%) and constant with time as previously published (supplemental Table 1).41 Due to the predominance of venous events, classical management with low-dose aspirin to prevent thrombosis should perhaps be questioned in this specific population as it mainly reduces the risk of arterial events. The use of anticoagulants could be a way to reduce the incidence of venous thrombotic events. In this cohort, 13.2% of patients received VKA and only 1.8% DOAC, all of them prescribed after a thrombotic event and not as primary prophylaxis. The role of DOACs is changing in the pediatric population as reflected by the start of the PREVAPIX-ALL trial, challenging apixaban vs placebo in primary prevention of thrombosis among children with acute lymphoblastic leukemia.43 Among arterial events, TIAs were the most frequent (44.8%), an intriguing finding as this diagnosis is sometimes difficult to make in adults, and we cannot exclude that some of them may have been misdiagnosed. Of note, classical risk factors (present in only 12.9% of patients) were not associated with arterial events in our cohort. In the logistic regression model, JAK2V617F mutation and hyperviscosity symptoms were significant risk factors for thrombosis risk in the whole cohort and for ET patients. For PV patients, none of the potential risk factors was confirmed in logistic regression model (Table 3). The prominence of hyperviscosity symptoms in this population is unique, perhaps reflecting microvascular occlusion, and merits further evaluation. In addition, presence of a JAK2 mutation impacts the median time to thrombosis in ET patients (28.5 years compared with not reached for all other patients, P = .02) (Figure 4) as in adult ET.44
A history of bleeding was found in 5.8% and new events in 9.5% of ET or PV cases (Table 2), which is lower than numbers observed in adults with ET or PV (12.5% and 15.3%, respectively).45 Here again, the cohort logistical regression model revealed that hyperviscosity symptoms (P = .015), splenomegaly (P = .026), bleeding history (P = .000), and platelet count > 1500 g/L (P = .034) were significant. Specifically for ET patients, hyperviscosity (P = .002), bleeding history (P < .001), platelet count > 1500 g/L (P = .002), and age < 20 years (P = .002) were all identified in the logistic regression model. Again, these are important findings and would support control of thrombocytosis even in young patients to reduce bleeding risk.
Myelofibrotic evolution was observed in 7.3% of ET and 8.8% of PV patients, respectively, comparable to the incidence observed in adults.17,46 This is in contrast with our previous review of the literature in young patients reporting low incidences of 1.8% and 2.7%, respectively.9 As >45% of events have been observed >10 years after diagnosis, the long follow-up of our cohort may explain these higher numbers, or perhaps better case ascertainment. An important difference with previous studies is that our cohort of patients has been collected from adult and not from pediatric centers, therefore allowing longer follow-up and appearance of those late transformation events. Secondary transformations to PV have been also observed in 5.8% of JAK2-mutated ET cases. Finally, transformations to AP/AML were anecdotal, despite the same long follow-up (Table 2). Here, logistical regression identified potential important new risks, including palpable splenomegaly, cardiovascular risk factors for ET patients; thrombosis history and age < 20 years for PV. Despite those high rates of complications, there was an excellent overall survival in this population, especially for ET and PV patients (Figure 3F). Adolescents and young adults have been previously identified to have better 5-year and 10-year survival rates than older patients.47
Currently, no prognostic scores have been specifically designed for or validated in young MPN patients (supplemental Tables 2 and 3). We assessed the prediction of survival and the risk of thrombosis utilizing current risk scores designed for adult MPN patients.3,16 -19 None of the scoring systems for survival performed well, and an important variability in results between IPSET-thrombosis and new IPSET-thrombosis scores was observed. This could relate to the impact of more advanced age and of prior thrombosis in current scores. Nevertheless, the incidence of thrombosis in our analysis during the follow-up was high (16.3% in PV and 10.1% in ET), and thus, prediction of this risk would be clinically useful.48-50 Leucocytosis was previously suggested to be an independent thrombosis risk factor in MPN patients, but in our study, a leukocyte count > 15 × 109/l was a trend predictive marker of thrombosis only in ET patients (P = .09).48,51 In the present analysis, cardiovascular risk factors were not frequent (12.9%, 56/434 patients), probably due to the young age, and played a minimal role in the development of thrombotic events. Correlations between JAK2V617F mutation and thrombotic complications were previously documented in MPN patients and could have an impact here, as could being linked with leucocytosis in ET.52,53 Next generation sequencing analyses are currently available in only 5.6% of the patients, but the collection of archival DNA has started to increase the number of patients with full clonal architecture that may give important information about survival and risks of transformation, thrombosis, or bleeding.54 Finally, recent data describing the occurrence of MPN driver mutations, sometimes many years before disease presentation, including sometimes in utero or during childhood, underpin a different biology of disease for this young cohort and may support a different weight in prognostic score.
The retrospective nature of this study could imply some biases. In particular, the number of patients included in this study does not necessarily reflect the total number of cases observed by country because members of the EHA MPN SWG, although reference centers in their country, are not the exclusive care centers for MPNs (supplemental Figure 1). In addition, most of the cases are diagnosed by pediatricians, and some of them may be missed in centers for adults. Recording of mutational status, absence of central review of bone marrow biopsies, and challenges in detecting splenomegaly are also pertinent. Furthermore, the cohort of patients included those diagnosed before 2000 (∼25%), when many institutions may not have performed bone marrow biopsy for diagnosis. We asked the clinicians to only include patients with a confirmed diagnosis, and thus, for these historical patients with long follow-up, any other causes, for example, of thrombocytosis, would have declared themselves. Finally, in the absence of specific guidelines for this population of young MPN patients, management could be heterogeneous among centers, potentially influencing the rate of complications. However, this is mitigated by the very high number of such extremely rare patients included in our cohort, suggesting that we revealed a reliable picture of MPN in very young patients.
Conclusion
We present the largest real-world study of young MPN patients to date, revealing some unexpected features. We observed a high disease burden, with incidences of thrombotic events and transformations higher than previously reported in this population even though many patients were being treated with cytoreductive agents. This study also provides new information for prospective biomarker studies (eg, histopathological and molecular analyses), clinical trials, and the development of specific treatment guidelines. We also demonstrate that current prognostic scores used in adult MPN do not perform well in this population and highlight novel risk factors such as hyperviscosity symptoms and splenomegaly.
Acknowledgments
The authors want to thank EHA SWG MPN group for its precious help to collect data and allow these analyses.
No funding was allocated to this study.
Authorship
Contribution: C.N.H., J.-J.K., and J.-C.I. conceived this study; Y.B., J.-C.I., and M. Sobas completed statistical analysis; J.-C.I., C.N.H., and M. Skowronek wrote the paper; C.N.H., and J.-J.K. reviewed the article; and all the authors contributed to data collection and approved the final version of the article.
Conflict-of-interest disclosure: M. Sobas reports advisory board membership (Novartis, BMS, Clegene) and speaker fees (Novartis, Abbvie). J.-J.K. reports advisory board membership (Novartis, AOP Orphan, BMS, Abbvie, Incyte) and speaker fees (Novartis, Pharmaessentia). M.F.M. reports advisory board membership (Novartis, BMS, Abbvie, Incyte, CTI, Sierra oncology), speaker bureau participation (Novartis, Abbvie, Pfizer, AOP, Incyte), and clinical trial support (BMS, AOP). J.G.T. reports advisory board membership (Novartis, BMS, Celgene, Pfizer) and honoraria (Novartis, BMS, Celgene, Abbvie, Pfizer). K. Laribi reports grants (Novartis, Takeda, Jansen, Abbvie) and personal fees (Novartis, Takeda, Abbvie, Iqone, Astellas, Astra, Beigene). K.L.D. reports honoraria (Abbvie, Incyte, Janssen, Roche,Takeda). M.G. reports consultancy (Amgen, AOP Orphan, Novartis, Celgene, CTI, Shire, Pfizer, Roche, Janssen, Gilead, Astra Zeneca) and honoraria (Amgen, AOP Orphan, Novartis, Celgene, CTI, Shire, Pfizer, Roche, Janssen, Gilead, Astra Zeneca). C.N.H. reports consultancy and honoraria (AOP Orphan, Novartis, Celgene, CTI, Roche, Janssen, Gilead, Geron, Galecto, Sierra), speaker bureau participation (AOP Orphan, Novartis, Celgene, CTI, Geron), and grants (Celgene, Constellation, Novartis). J.-C.I. reports grants, honoraria, and advisory board membership (Novartis). The remaining authors declare no competing financial interests.
Correspondence: Jean-Christophe Ianotto, Service d’Hématologie Clinique, Institut de Cancéro-Hématologie, Hopital Morvan, Avenue Foch, CHRU de Brest, 29609 Brest Cedex, France; e-mail: jean-christophe.ianotto@chu-brest.fr.

