

Key Points
VWD type 2 is the result of variants that lead to dysfunctional VWF.
The vast majority of patients included in this large cohort (88%) had missense variants.

Abstract
von Willebrand disease (VWD) type 2 is caused by qualitative abnormalities of von Willebrand factor (VWF). This study aimed to determine the genotypic and phenotypic characterizations of a large VWD type 2 cohort from Milan. We included 321 patients (54% female) within 148 unrelated families from 1995 to 2021. Patients were fully characterized using laboratory phenotypic tests, and the genotypic diagnosis was confirmed by target genetic analysis using Sanger sequencing. Patients were diagnosed with type 2A (n = 98; 48 families), 2B (n = 85; 38 families), 2M (n = 112; 50 families), or 2N (n = 26; 12 families). Eighty-two unique VWF variants, including 8 novel variants, were found. The potential pathogenic effect of novel variants was assessed by in silico analysis. Most patients were heterozygous for a single variant (n = 259; 81%), whereas 37 cases (11%) had 2 variants (4 homozygous, 9 in trans, and 24 in cis). Twenty-five patients (8%) had ≥3 variants, mainly as a result of gene conversions. Among the 82 distinct variants identified, 5 different types, including missense (n = 64), gene conversion (n = 10), synonymous (n = 1), deletion (n = 4), and splice (n = 3), were observed. The results from this large cohort showed that VWD type 2 is invariably due to variants that do not prevent the synthesis of the protein, and a vast majority of patients (88%) had missense variants. Given the complexity of type 2 diagnosis and the necessity of performing several phenotypic tests, genetic analysis for patients suspected of having type 2 is beneficial to establish the correct diagnosis.
Introduction
von Willebrand disease (VWD), with an estimated prevalence of about 1% in the general population, is the most common congenital bleeding disorder1,2 that results from quantitative or qualitative defects in the von Willebrand factor (VWF) glycoprotein.3 Quantitative reductions include type 1 with a partial decrease in VWF and type 3 with a complete absence. Qualitative abnormalities cause type 2, which is further divided into 4 groups, including type 2A, 2B, 2M, and 2N.4
As an adhesive glycoprotein, VWF supports platelet adhesion/aggregation at sites of vascular damage under high shear stress conditions and protects the procoagulant factor VIII (FVIII), delivering it to the sites where it is needed.5,6 VWF is synthesized in the endothelial cells and megakaryocytes as a pre-pro-protein of 2813 amino acids, with domains orderly arranged in D1–D2–D′–D3–A1–A2–A3–D4–C1–C2–C3–C4–C5–C6–CK. The D1-D2 domain (propeptide) is cleaved by furin and produces a mature VWF subunit.6,7 VWF multimers with a molecular weight of >20 000 kDa are formed by CK domain dimerization in the endoplasmic reticulum and disulfide bonding in the Golgi.6,7
VWD type 2A is characterized by a reduction in platelet adhesion resulting from either loss or reduction of large VWF multimers or defective synthesis of VWF multimers. The different pathologic mechanisms and the corresponding VWD types include increased proteolysis by thrombospondin type 1 motif member 13 (ADAMTS-13) in the A2 domain (IIA), multimerization defects in the D1-D2 domain with reduced proteolytic cleavage (IIC), dimerization defects in the cystine knot (IID), and impaired VWF multimerization with reduced proteolytic cleavage in the D3 domain (IIE).8,9 In type 2B, the mutated VWF A1 domain has an enhanced affinity for the platelet GPIb, leading to some degree of loss of the largest multimers and occasionally decreased platelet count.10 However, type 2B Malmo/New York (2B NY), which is often due to conversions between the VWF gene (VWF) and its pseudogene, presents with normal multimers and platelet count.11,12 Type 2M is characterized by a reduced affinity of the A1 domain for GPIb (classical 2M).4 The decreased affinity of VWF for collagen, resulting from variants in either the A1 or A3 domain, has also been described in type 2M (2MCB).13 Gene variants located in the D′-D3 domains, which compromise the binding of FVIII, cause type 2N.14
All VWD type 2 are dominantly inherited, with the exception of type 2A(IIC) and type 2N, which are recessive.8 At variance with VWD type 1, VWF gene variants are generally fully penetrant in type 2A, 2B, and 2M.15 Genetic testing in type 2 is more straightforward than in type 1 or 3, because most of the variants are located in peculiar VWF functional domains.15 Furthermore, when some biochemical tests, such as multimer analysis, ristocetin-induced platelet agglutination (RIPA), and FVIII-binding activity of VWF (VWF:FVIIIB), are not available, genetic analysis can be a good surrogate to establish the diagnosis.
Because a small number of studies have comprehensively investigated the genetic basis of VWD type 2, we aimed to evaluate the laboratory phenotypic and genotypic characterizations and the variant profile of a large VWD type 2 cohort from Milan.
Patients and methods
Patients
We included all genetically confirmed patients with VWD type 2 diagnosis referred to the A. Bianchi Bonomi Hemophilia and Thrombosis Center in Milan from 1995 to 2021. The institutional review board of the Fondazione Istituto di Ricovero e Cura a Carattere Scientifico Ca’ Granda Ospedale Maggiore Policlinico approved the study. Informed consent was obtained from all patients according to the Declaration of Helsinki. All patients were extensively investigated for clinical manifestation and VWD biochemical phenotypic tests to achieve the diagnosis. Their classification was established according to the International Society on Thrombosis and Haemostasis Scientific and Standardization Committee guidelines.4
Laboratory evaluation
Blood sample collection from each patient was carried out at the time of study enrollment. For the biochemical measurements, blood samples were collected in a 3.2% buffered citrate solution (1:9 ratio of anticoagulant/whole blood), centrifuged at 1500g for 15 minutes at room temperature, and then aliquoted and stored at −80°C until the date of assessment. Because the laboratory measurements were performed over a time period from 1995 to 2021, different methods were used to evaluate the VWF level and its activities in this period (data supplement), as previously described by Biguzzi et al.16 In brief, the VWF antigen (VWF:Ag) was measured by enzyme-linked immunosorbent assay or immunoturbidometric assay (HemosIL VWF:Ag). VWF platelet–dependent activity (referred to as VWF activity) was measured using aggregometric or automated coagulometer (described as VWF ristocetin cofactor activity), VWF monoclonal antibody activity (HemosIL VWF activity), or VWF:GPIbR (HemosIL VWF ristocetin cofactor activity) methods. FVIII clotting activity (FVIII:C) is always assessed by a 1-stage assay. VWF activity, VWF:Ag, and FVIII:C were evaluated as the first-level tests. If patients showed reduced levels or had a low ratio of VWF activity/VWF:Ag, we further performed VWF collagen binding (VWF:CB), with collagen type I or III or a combination of thereof, RIPA and VWF multimer analysis assays. In cases with a low FVIII:C/VWF:Ag ratio, VWF:FVIIIB was performed to confirm or exclude a type 2N diagnosis.
Genotypic analysis
Genetic analysis was performed for all patients using polymerase chain reaction and Sanger sequencing. The target sequencing approach was carried out to find the candidate variants by amplifying the exon encoding for specific VWF domains including intron-exon boundaries. Variants are reported following the guidelines of the Human Genome Variation Society.17 The Human Gene Mutation Database18 and the Leiden Open Variation Database19 were used to understand the already reported genotypic-phenotypic association of variants.
In silico analysis
In silico prediction was assessed to evaluate the functional effects of the novel variants by Polymorphism Phenotyping (PolyPhen; http://genetics.bwh.harvard.edu/pph2/), Sorting Intolerant From Tolerant (SIFT; http://provean.jcvi.org/), Combined Annotation Dependent Depletion (CADD; https://cadd.gs.washington.edu/), Protein Variation Effect Analyzer (PROVEAN; http://provean.jcvi.org/index.php/), Mendelian Clinically Applicable Pathogenicity (M-CAP; http://bejerano.stanford.edu/MCAP/), and VarSome (https://varsome.com/) software classification. All the aforementioned websites have been accessed in December 2021.
Results
Patients and families
A total of 321 patients (174 female and 147 male) with a diagnosis of VWD type 2 from 148 families were included. The mean number of individuals per family was 2.2, and 1-generation family (75 families) was the most common family structure. Forty-six families were 2-generations, and 25 were 3-generations. Only 1 family had a 4-generation structure.
The phenotypic data for all patients are summarized in Table 1. Ninety-eight patients (48 families) were diagnosed with type 2A. They had the lowest overall median values for VWF activity, VWF activity/VWF:Ag ratio, VWF:CB, and VWF:CB/VWF:Ag ratio among all those with type 2 (Table 1). There were 85 patients (38 families) with type 2B. One hundred twelve patients (50 families) were classified as having type 2M, and overall, they had the lowest median values for VWF:Ag and VWF activity (similar to type 2A) among all types. Twenty-six patients with type 2N (12 families) presented with reduced VWF:FVIIIB activity, and 16 of them had markedly reduced binding, with a median FVIII:C of 37 IU/dL, and 10 had mildly reduced binding, with a median FVIII:C of 71 IU/dL.
General characterization and laboratory results of patients with VWD type 2 in the Milan cohort
| VWD type | N of patients | FVIII:C, IU/dL−1 | VWF:Ag, IU/dL−1 | VWF activity, IU/dL−1 | VWF activity/VWF:Ag ratio | VWF:CB, IU/dL−1* | VWF:CB/VWF:Ag ratio |
|---|---|---|---|---|---|---|---|
| All 2A | 98 | 47 (20-196) | 33 (7-370) | 11 (5-88) | 0.37 (0.06-1) | 7 (1-59) | 0.2 (0.02-0.9) |
| 2A(IIA) | 64 | 52 (20-100) | 45 (7-130) | 10 (4-29) | 0.28 (0.06-0.68) | 6 (1-26) | 0.15 (0.02-0.7) |
| 2A(IIE) | 32 | 40 (24-96) | 20 (13-149) | 13 (6-60) | 0.52 (0.23-1) | 10 (4-41) | 0.41 (0.2-0.9) |
| 2A(IIC) | 1 | 56 | 18 | 8 | 0.44 | 3 | 0.17 |
| 2A(IIH) | 1 | 196 | 370 | 88 | 0.23 | 59 | 0.16 |
| All 2B | 85 | 62 (6-132) | 55 (18-170) | 25 (4-99) | 0.46 (0.1-1) | 21 (2-82) | 0.4 (0.06-1.4) |
| Classical 2B | 64 | 61 (6-132) | 52 (18-170) | 22 (4-99) | 0.35 (0.1-0.93) | 15 (2-62) | 0.2 (0.06-1) |
| 2B NY | 21 | 70 (40-105) | 57 (21-80) | 39 (10-73) | 0.7 (0.4-1) | 45 (7-82) | 0.87 (0.33-1.4) |
| All 2M† | 112 | 50 (23-133) | 28 (12-147) | 11 (5-101) | 0.47 (0.11-1.1) | 15 (3-80) | 0.58 (0.1-1.6) |
| Classical 2M | 46 | 48 (23-124) | 26 (12-130) | 11 (5-44) | 0.42 (0.14-1) | 17 (8-80) | 0.7 (0.3-1.3) |
| 2M/2A | 37 | 48 (24-133) | 29 (12-90) | 9 (5-23) | 0.29 (0.15-0.68) | 14 (3-52) | 0.5 (0.1-0.95) |
| 2MCB | 29 | 51 (35-133) | 30 (18-147) | 25 (11-101) | 0.8 (0.5-1) | 14 (7-66) | 0.5 (0.14-1) |
| All 2N‡ | 26 | 45 (13-102) | 69 (22-156) | 60 (18-144) | 0.85 (0.68-1.3) | 53 (12-134) | 0.87 (0.5-1.4) |
| 2N | 16 | 37 (13-50) | 60 (22-156) | 41 (18-106) | 0.8 (0.63-1.40) | 39 (12-61) | 0.88 (0.5-1.4) |
| Carrier 2N | 10 | 71 (50-102) | 98 (56-121) | 88 (47-144) | 0.89 (0.64-1.1) | 72 (59-88) | 0.97 (0.72-1.2) |
| VWD type | N of patients | FVIII:C, IU/dL−1 | VWF:Ag, IU/dL−1 | VWF activity, IU/dL−1 | VWF activity/VWF:Ag ratio | VWF:CB, IU/dL−1* | VWF:CB/VWF:Ag ratio |
|---|---|---|---|---|---|---|---|
| All 2A | 98 | 47 (20-196) | 33 (7-370) | 11 (5-88) | 0.37 (0.06-1) | 7 (1-59) | 0.2 (0.02-0.9) |
| 2A(IIA) | 64 | 52 (20-100) | 45 (7-130) | 10 (4-29) | 0.28 (0.06-0.68) | 6 (1-26) | 0.15 (0.02-0.7) |
| 2A(IIE) | 32 | 40 (24-96) | 20 (13-149) | 13 (6-60) | 0.52 (0.23-1) | 10 (4-41) | 0.41 (0.2-0.9) |
| 2A(IIC) | 1 | 56 | 18 | 8 | 0.44 | 3 | 0.17 |
| 2A(IIH) | 1 | 196 | 370 | 88 | 0.23 | 59 | 0.16 |
| All 2B | 85 | 62 (6-132) | 55 (18-170) | 25 (4-99) | 0.46 (0.1-1) | 21 (2-82) | 0.4 (0.06-1.4) |
| Classical 2B | 64 | 61 (6-132) | 52 (18-170) | 22 (4-99) | 0.35 (0.1-0.93) | 15 (2-62) | 0.2 (0.06-1) |
| 2B NY | 21 | 70 (40-105) | 57 (21-80) | 39 (10-73) | 0.7 (0.4-1) | 45 (7-82) | 0.87 (0.33-1.4) |
| All 2M† | 112 | 50 (23-133) | 28 (12-147) | 11 (5-101) | 0.47 (0.11-1.1) | 15 (3-80) | 0.58 (0.1-1.6) |
| Classical 2M | 46 | 48 (23-124) | 26 (12-130) | 11 (5-44) | 0.42 (0.14-1) | 17 (8-80) | 0.7 (0.3-1.3) |
| 2M/2A | 37 | 48 (24-133) | 29 (12-90) | 9 (5-23) | 0.29 (0.15-0.68) | 14 (3-52) | 0.5 (0.1-0.95) |
| 2MCB | 29 | 51 (35-133) | 30 (18-147) | 25 (11-101) | 0.8 (0.5-1) | 14 (7-66) | 0.5 (0.14-1) |
| All 2N‡ | 26 | 45 (13-102) | 69 (22-156) | 60 (18-144) | 0.85 (0.68-1.3) | 53 (12-134) | 0.87 (0.5-1.4) |
| 2N | 16 | 37 (13-50) | 60 (22-156) | 41 (18-106) | 0.8 (0.63-1.40) | 39 (12-61) | 0.88 (0.5-1.4) |
| Carrier 2N | 10 | 71 (50-102) | 98 (56-121) | 88 (47-144) | 0.89 (0.64-1.1) | 72 (59-88) | 0.97 (0.72-1.2) |
Data are presented as median (range).
Missing number for VWF:CB in type 2A (n = 11), 2B (n = 5), 2M (n = 7), and 2N (n = 11).
Type 2M could be due to distinct etiologies, either a defective binding of VWF to platelet GPIb (classical 2M) or collagen (2MCB). Patients with classical 2M had a reduced VWF activity/VWF:Ag ratio with a normal median VWF:CB/VWF:Ag ratio, whereas in 2MCB, the opposite is actually the case. For this reason, the VWF:CB/VWF:Ag ratio of 2M, which represents all three subgroup, is 0.58, whereas classical 2M has a ratio of 0.7.
Type 2N includes patients who showed a markedly (2N) or slightly (carrier 2N) reduced VWF:FVIIIB result.
Gene variants
A total number of 82 different variants, including 8 novel , were identified. Although several nucleotide changes were presented in most gene conversions, each was counted as a single variant.
The list of identified variants and their locations on the pro-VWF is depicted in Figure 1. Most patients were heterozygotes for a single variant (n = 259; 81%), whereas 37 (11%) had 2 variants. Among the last group, 4 patients (1%) were homozygotes (type 2N), 9 (3%) were compound heterozygotes, and 24 (7%) had nucleotide changes in the same allele (16 nongene conversions and 8 gene conversions). Twenty-five patients (8%) had ≥3 variants, mainly as a result of gene conversions.

Variants identified in 321 patients with VWD type 2. Eighty-two unique variants, including 8 novel variants, were found. A vast majority (78%) of identified variants were missense substitutions, although other types, such as gene conversions (12%), synonymous variants (1%), splice variants (4%), and deletions (5%), were also observed. Nevertheless, in dominant VWD types (2A, 2B, and 2M), all variants identified always led to the synthesis of mutated VWF. Most patients were heterozygotes for a single variant (n = 259; 81%), whereas 37 patients (11%) had 2 variants: 4 (1%) were homozygotes, and 9 (3%) were in trans and 24 (7%) in cis position. Twenty-five patients (8%) had ≥3 variants, mainly as a result of gene conversion. #Six distinct gene conversions in type 2B NY were determined, and p.Pro1266Leu was the core variant.
Variants identified in 321 patients with VWD type 2. Eighty-two unique variants, including 8 novel variants, were found. A vast majority (78%) of identified variants were missense substitutions, although other types, such as gene conversions (12%), synonymous variants (1%), splice variants (4%), and deletions (5%), were also observed. Nevertheless, in dominant VWD types (2A, 2B, and 2M), all variants identified always led to the synthesis of mutated VWF. Most patients were heterozygotes for a single variant (n = 259; 81%), whereas 37 patients (11%) had 2 variants: 4 (1%) were homozygotes, and 9 (3%) were in trans and 24 (7%) in cis position. Twenty-five patients (8%) had ≥3 variants, mainly as a result of gene conversion. #Six distinct gene conversions in type 2B NY were determined, and p.Pro1266Leu was the core variant.
Among the 82 unique variants identified, 5 different types were found (Figure 2). Missense variants were largely the most common (n = 64; 78%). Gene conversions (n = 10) comprised 12% of variants and were found in type 2B NY or classical type 2M. A synonymous variant (1%) was found in patients with type 2A(IIE) (c.3390C>T, p.Cys1130Cys causing p.Pro1127_Gly1180delinsArg). Three distinct splice variants (4%), 1 in type 2N (c.2546 + 3G>C/p.Arg854Gln) and 2 in type 2A(IIE) (c.3108 + 5G>A and c.3380-2A>G causing p.Pro1127_Gly1180delinsArg), were observed. Four different small deletions (5%) were also detected: 1 out-of-frame deletion in type 2A(IIC) (c.1092_1093delTC/c.1583A>G, p.Asp366Leufs*16/p.Asn528Ser) and 3 in-frame deletions (1 in type 2A(IIA) [c.4606_4611delCACGTC, p.His1536_Val1537del] and 2 in type 2M [c.4222_4224delAAG, p.Lys1408del and c.3831_3833delCCT, p.Asp1277_Leu1278delinsGlu]).
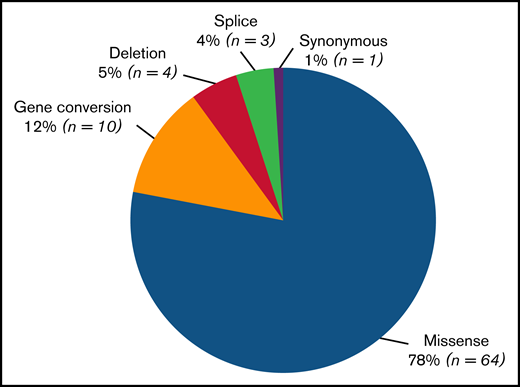
Number of different variants types found in the VWD type 2 Milan cohort. Eighty-two unique variants, including 8 novel variants, were identified. The most frequent type of variant identified was missense substitution (n = 64; 78%), followed by gene conversions (n = 10; 12%), synonymous variants (n = 1; 1.5%), splice variants (n = 3; 3.5%), and deletions (n = 4, 5%). Of 4 different small deletions detected, 1 was out of frame and was found in type 2A(IIC) (c.1092_1093delTC/c.1583A>G, p.Asp366Leufs*16/p.Asn528Ser), and 3 were in frame, 1 in type 2A(IIA) (c.4606_4611delCACGTC, p.His1536_Val1537del)52 and 2 in type 2M (c.4222_4224delAAG, p.Lys1408del and c.3831_3833delCCT, p.Asp1277_Leu1278delinsGlu).
Number of different variants types found in the VWD type 2 Milan cohort. Eighty-two unique variants, including 8 novel variants, were identified. The most frequent type of variant identified was missense substitution (n = 64; 78%), followed by gene conversions (n = 10; 12%), synonymous variants (n = 1; 1.5%), splice variants (n = 3; 3.5%), and deletions (n = 4, 5%). Of 4 different small deletions detected, 1 was out of frame and was found in type 2A(IIC) (c.1092_1093delTC/c.1583A>G, p.Asp366Leufs*16/p.Asn528Ser), and 3 were in frame, 1 in type 2A(IIA) (c.4606_4611delCACGTC, p.His1536_Val1537del)52 and 2 in type 2M (c.4222_4224delAAG, p.Lys1408del and c.3831_3833delCCT, p.Asp1277_Leu1278delinsGlu).
In 6 type 2N patients, despite markedly reduced VWF:FVIIIB and FVIII:C and/or reduced level of VWF, suggesting the likelihood of a second VWF variant, we found only p.Arg854Gln in the heterozygous state. However, because of the reduced VWF:FVIIIB levels at the time of diagnosis, we only sequenced the coding region of D′-D3 domains. Unfortunately, samples of the aforementioned patients are no longer available for further investigation, so the second expected variants remain to be identified.
Prevalence
To evaluate the prevalence of each VWD type 2, we considered all 321 patients. Type 2M was the most common form (35%), followed by type 2A (31%), type 2B (26%), and type 2N (8%). A similar pattern was observed when only the index cases (n = 148 families) were considered: type 2M (34%), 2A (32%), 2B (26%), and 2N (8%).
We further subclassified type 2A patients into 2A(IIA), 2A(IIE), 2A(IIC), and 2A(IIH); type 2B in classical 2B and 2B NY; type 2M in classical 2M, 2M/2A, and 2MCB; and type 2N in 2N and 2N carriers. This classification was based on the combined evaluation of the laboratory phenotypic and genotypic results. Figure 3 shows the prevalence of all type 2 subgroups in the Milan cohort.
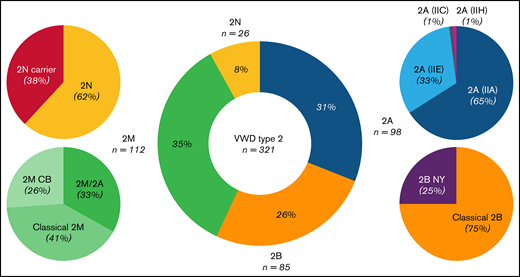
Frequency spectrum of 321 patients diagnosed with VWD type 2 in the Milan cohort study. Type 2M was the most common type (35%), followed by 2A (31%), 2B (26%), and 2N (8%). In type 2M, 41% of patients had variants at the VWF A1 domain, with a platelet-binding defect (classical 2M), whereas 26% had variants at the A3 domain, with a collagen-binding defect (2MCB). Almost one third of type 2M patients (33%), with variants in the A1 domain, shared a similar phenotype between type 2M and 2A, and they were classified as 2M/2A. In type 2A, 65% of patients had variants located at the A2 domain, with an enhanced susceptibility for ADAMTS-13, and were classified as 2A(IIA), with an exceptional case in the A1 domain. The remaining 33% were located at the D3 domain, with multimerization defects, and were classified as 2A(IIE). Patients with a diagnosis of type 2A(IIC) or 2A(IIH) made up only 2% of the type 2A cohorts. Most patients with 2B (75%) had variants at the A1 domain, with a platelet-binding defect (classical 2B), and the rest (25%) had variants at the D3-A1 junction, classified as 2B NY. Patients with 2N were mainly compound heterozygotes for 2N/type 1 or 3 (46%), and only 16% were homozygotes for the 2N variant. Type 2N carriers accounted for 38% of the 2N population.
Frequency spectrum of 321 patients diagnosed with VWD type 2 in the Milan cohort study. Type 2M was the most common type (35%), followed by 2A (31%), 2B (26%), and 2N (8%). In type 2M, 41% of patients had variants at the VWF A1 domain, with a platelet-binding defect (classical 2M), whereas 26% had variants at the A3 domain, with a collagen-binding defect (2MCB). Almost one third of type 2M patients (33%), with variants in the A1 domain, shared a similar phenotype between type 2M and 2A, and they were classified as 2M/2A. In type 2A, 65% of patients had variants located at the A2 domain, with an enhanced susceptibility for ADAMTS-13, and were classified as 2A(IIA), with an exceptional case in the A1 domain. The remaining 33% were located at the D3 domain, with multimerization defects, and were classified as 2A(IIE). Patients with a diagnosis of type 2A(IIC) or 2A(IIH) made up only 2% of the type 2A cohorts. Most patients with 2B (75%) had variants at the A1 domain, with a platelet-binding defect (classical 2B), and the rest (25%) had variants at the D3-A1 junction, classified as 2B NY. Patients with 2N were mainly compound heterozygotes for 2N/type 1 or 3 (46%), and only 16% were homozygotes for the 2N variant. Type 2N carriers accounted for 38% of the 2N population.
Common variants
Only the index cases of each family were considered, with the goal of evaluating the most commonly identified variants (Table 2). In type 2A(IIA), p.IIe1628Thr was observed in 7 (21%) of 34 families. Two other variants were also recurrent (p.Ser1506Leu and p.Arg1597Gln); each was identified in 4 (12%) of 34 families. A synonymous variant (c.3390C>T, p.Cys1130Cys causing p.Pro1127_Gly1180delinsArg) was identified in 4 (33%) of the 12 families with type 2A(IIE) variants. In classical type 2B, variants p.Arg1308Cys (16%), p.Arg1341Trp (16%), and p.Val1316Met (13%) were relatively common among the 31 families investigated. In type 2B NY, 6 distinct gene conversions were found, and all patients shared a substitution of proline 1266. Among 24 families with classical 2M, p.Ala1377Val-Arg1379Cys variants were found in 4 (17%). In the 2M/2A group (16 families), p.Arg1374His (69%) and p.Arg1374Cys (25%) were the prevalent variants. p.Ala1716Pro was the most common variant in 2MCB, found in 4 (40%) of 10 families. In type 2N, all 12 families had p.Arg854Gln, either in homozygosity, heterozygosity (carrier), or compound heterozygosity with VWF quantitative variants.
Most common variants identified in VWF for each VWD type 2 in the index cases
| VWD type | Common variants in index cases (n; %) |
|---|---|
| 2A(IIA) | p.Ile1628Thr (7; 21) |
| Index case (n = 33) | p.Ser1506Leu (4; 12) |
| All 2A(IIA) patients (n = 64) | p.Arg1597Gln (4; 12) |
| 2A(IIE) | c.3390C>T, p.Cys1130Cys (4; 33)* |
| Index case (n = 12) | |
| All 2A(IIE) patients (n = 32) | |
| Classical 2B | p.Arg1308Cys (5; 16) |
| Index case (n = 31) | p.Arg1341Trp (5; 16) |
| All classical 2B patients (n = 61) | p.Val1316Met (4; 13) |
| 2B NY | p.Pro1266Leu† |
| Index case (n = 7) | |
| All 2B NY patients (n = 24) | |
| Classical 2M | p.Ala1377Val-Arg1379Cys (4; 17)‡ |
| Index case (n = 24) | p.Arg1399Cys (2; 8) |
| All classical 2M patients (n = 46) | |
| 2M/2A | p.Arg1374His (11; 69) |
| Index case (n = 16) | p.Arg1374Cys (4; 25) |
| All 2M/2A patients (n = 37) | |
| 2MCB | p.Ala1716Pro (4; 40) |
| Index case (n = 10) | p.Tyr1780Asn (2; 20) |
| All 2MCB patients (n = 29) | |
| 2N | p.Arg854Gln§ |
| Index case (n = 12) | |
| All 2N patients (n = 26) |
| VWD type | Common variants in index cases (n; %) |
|---|---|
| 2A(IIA) | p.Ile1628Thr (7; 21) |
| Index case (n = 33) | p.Ser1506Leu (4; 12) |
| All 2A(IIA) patients (n = 64) | p.Arg1597Gln (4; 12) |
| 2A(IIE) | c.3390C>T, p.Cys1130Cys (4; 33)* |
| Index case (n = 12) | |
| All 2A(IIE) patients (n = 32) | |
| Classical 2B | p.Arg1308Cys (5; 16) |
| Index case (n = 31) | p.Arg1341Trp (5; 16) |
| All classical 2B patients (n = 61) | p.Val1316Met (4; 13) |
| 2B NY | p.Pro1266Leu† |
| Index case (n = 7) | |
| All 2B NY patients (n = 24) | |
| Classical 2M | p.Ala1377Val-Arg1379Cys (4; 17)‡ |
| Index case (n = 24) | p.Arg1399Cys (2; 8) |
| All classical 2M patients (n = 46) | |
| 2M/2A | p.Arg1374His (11; 69) |
| Index case (n = 16) | p.Arg1374Cys (4; 25) |
| All 2M/2A patients (n = 37) | |
| 2MCB | p.Ala1716Pro (4; 40) |
| Index case (n = 10) | p.Tyr1780Asn (2; 20) |
| All 2MCB patients (n = 29) | |
| 2N | p.Arg854Gln§ |
| Index case (n = 12) | |
| All 2N patients (n = 26) |
The apparent synonymous variant is responsible for the skipping of exon 26, which results in a large in-frame deletion (p.P1127_G1180delinsR).34
Six different gene conversions from 2 up to 5 nuclide changes, always with p.Pro1266Lue, were identified.
These 2 variants have been found in the same allele, and their combination is responsible for the classical 2M phenotype.35
p.Arg854Gln was found in type 2N patients, either in homozygosity or compound heterozygosity, and carriers.
Cases with multiple variants
In 61 patients (28 families), more than one nucleotide change in VWF was found. Thirty patients had gene conversions, whereas in the remaining 31 patients, 2 or 3 variants were identified in either trans or cis position (supplemental Table 1). In the latter group, variants resulted in type 2A(IIC) (p.Asp366Leufs*16/p.Asn528Ser), type 2A(IIH) (p.Arg202Trp-Arg1583Gln/p.Cys849Tyr), classical 2B (p.Cys275Arg/p.Pro1337Leu and p.Arg1308Cys/p.Gly1172Val), classical 2M (p.Ala1377Val-p.Arg1379Cys), 2M/2A (p.Arg924Gln-p.Arg1315Leu), and 2N (p.Arg854Gln/p.Leu893Arg, p.Arg854Gln/p.Arg854Gln, p.Arg854Gln/p.Tyr1584Cys, and p.Arg854Gln/c.2546 + 3G>C). Gene conversions, occurring from 2 up to 5 nucleotide changes, were found in type 2B NY and classical 2M (supplemental Table 2). Ten unique gene conversions (6 in 2B NY and 4 in classical 2M) were identified in 30 patients within 12 families. Two patients with gene conversions carried also other variants in trans (p.Ser1263Ser-Pro1266Leu/p.Cys2557Tyr and p.Pro1266Leu-Val1279Ile/p.Ala1377Val-R1379Cys).
Novel variants and in silico prediction
We found 8 different novel candidate variants in VWF (p.Leu893Arg, p.Cys1126Tyr, p.Cys1142Phe, p.Leu1281Arg, p.Arg1426Pro, p.Leu1657Pro, p.Ser1731Leu, and p.Cys2557Tyr). All variants were missense and were located at the mature VWF subunit. Using 5 different in silico prediction tools (PolyPhen, SIFT, CADD, M-CAP, and PROVEAN) and VarSome classification, the functional effects of these variants were assessed (Table 3). All missense variants were predicted as probably pathogenic by SIFT, CADD, M-CAP, and PROVEAN tools. The VarSome algorithm classified 4 variants as likely pathogenic (p.Leu1281Arg, p.Arg1426Pro, p.Leu1657Pro, and p.Ser1731Leu), whereas the other 4 were classified as variants of uncertain significance (Table 3).
Genotypic-phenotypic characterization and in silico predication of novel variants in the index cases with VWD type 2
| Family | Nucleotide change, amino acid change | Exon | FVIII:C, IU/dL−1 | VWF:Ag, IU/dL−1 | VWF activity, IU/dL−1 | VWF:CB, IU/dL−1 | VWF activity/VWF:Ag ratio | Diagnosis | In silico analysis* | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PolyPhen | SIFT | CADD | M-CAP | PROVEAN | VarSome | |||||||||
| I | c.2678T>G, p.Leu893Arg | 20 | 28 | 30 | 29 | 27 | 0.96 | 2N; compound with p.R854Q in trans | 1 | 0 | 31 | 0.435 | −5.78 | VUS |
| II | c.3377G>A, p.Cys1126Tyr | 25 | 40 | 19 | 10 | 10 | 0.52 | 2A(IIE) | 0.89 | 0 | 25.3 | 0.89 | −10.8 | VUS |
| III | c.3425G>T, p.Cys1142Phe | 26 | 71 | 105 | 42 | 34 | 0.4 | 2A(IIE) | 1 | 0 | 27.3 | 0.18 | −10.54 | VUS |
| IV | c.3842T>G, p.Leu1281Arg | 28 | 26 | 14 | 6 | 9 | 0.42 | 2A(IIE) | 1 | 0 | 27.5 | 0.75 | −2.08 | Likely pathogenic |
| V | c.4277G>C, p.Arg1426Phe | 28 | 64 | 39 | 12 | 31 | 0.31 | 2M | 0.81 | 0 | 23.6 | 0.72 | −1.35 | Likely pathogenic |
| VI | c.4970T>C, p.Leu1657Phe | 28 | 51 | 35 | 8 | 2 | 0.22 | 2A(IIA) | 1 | 0 | 24.7 | 0.84 | −2.82 | Likely pathogenic |
| VII | c.5192C>T, p.Ser1731Leu | 30 | 74 | 67 | 41 | 12 | 0.61 | 2MCB | 0.78 | 0 | 28.4 | 0.67 | −3.2 | Likely pathogenic |
| VIII | c.7670G>A, p.Cys2557Tyr | 45 | 58 | 21 | 10 | 7 | 0.47 | 2B NY: compound with p.S1263S-P1266L in trans | 1 | 0 | 24.2 | 0.22 | −4.94 | VUS |
| Family | Nucleotide change, amino acid change | Exon | FVIII:C, IU/dL−1 | VWF:Ag, IU/dL−1 | VWF activity, IU/dL−1 | VWF:CB, IU/dL−1 | VWF activity/VWF:Ag ratio | Diagnosis | In silico analysis* | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PolyPhen | SIFT | CADD | M-CAP | PROVEAN | VarSome | |||||||||
| I | c.2678T>G, p.Leu893Arg | 20 | 28 | 30 | 29 | 27 | 0.96 | 2N; compound with p.R854Q in trans | 1 | 0 | 31 | 0.435 | −5.78 | VUS |
| II | c.3377G>A, p.Cys1126Tyr | 25 | 40 | 19 | 10 | 10 | 0.52 | 2A(IIE) | 0.89 | 0 | 25.3 | 0.89 | −10.8 | VUS |
| III | c.3425G>T, p.Cys1142Phe | 26 | 71 | 105 | 42 | 34 | 0.4 | 2A(IIE) | 1 | 0 | 27.3 | 0.18 | −10.54 | VUS |
| IV | c.3842T>G, p.Leu1281Arg | 28 | 26 | 14 | 6 | 9 | 0.42 | 2A(IIE) | 1 | 0 | 27.5 | 0.75 | −2.08 | Likely pathogenic |
| V | c.4277G>C, p.Arg1426Phe | 28 | 64 | 39 | 12 | 31 | 0.31 | 2M | 0.81 | 0 | 23.6 | 0.72 | −1.35 | Likely pathogenic |
| VI | c.4970T>C, p.Leu1657Phe | 28 | 51 | 35 | 8 | 2 | 0.22 | 2A(IIA) | 1 | 0 | 24.7 | 0.84 | −2.82 | Likely pathogenic |
| VII | c.5192C>T, p.Ser1731Leu | 30 | 74 | 67 | 41 | 12 | 0.61 | 2MCB | 0.78 | 0 | 28.4 | 0.67 | −3.2 | Likely pathogenic |
| VIII | c.7670G>A, p.Cys2557Tyr | 45 | 58 | 21 | 10 | 7 | 0.47 | 2B NY: compound with p.S1263S-P1266L in trans | 1 | 0 | 24.2 | 0.22 | −4.94 | VUS |
VUS, variant of uncertain (or unknown) significance.
Polyphen score: >0.908, probably damaging; >0.446 and ≤0.908, possibly damaging; ≤0.446, benign. SIFT score: <0.05, deleterious; ≥0.05, tolerated. CADD score: >20, deleterious. M-CAP: >0.025, pathogenic. PROVEAN: ≤−2.5, deleterious; >−2.5, neutral.
Cases with discrepancies for VWD classification
Genetic testing in VWD type 2 helps to confirm the diagnosis. Nevertheless, in the literature, several variants have been ascribed to different VWD types, so they are of difficult categorization within a specific type. In our study, these variants included p.Tyr1107Cys, p.Cys1130Phe, p.Tyr1146Cys, p.Arg1315Cys, p.Arg1315Leu, p.Arg1374His, p.Arg1374Cys, p.Arg1399Cys, p.Ala1716Pro, and p.Val1820Glu (Table 4). p.Tyr1107Cys, p.Cys1130Phe, and p.Tyr1146Cys were classified as type 2A(IIE) according to their multimer patterns, because an intermediate-resolution gel showed the absence of satellite bands in the triplet structure. Variants p.Arg1374His, p.Arg1374Cys, and p.Arg1315Leu were diagnosed as type 2M/2A because of severely decreased VWF activity/VWF:Ag and mildly decreased VWF:CB/VWF:Ag ratios along with a minor loss of large VWF multimers (Table 4; supplemental Figure 1). A normal VWF:CB/VWF:Ag ratio, as well as a normal multimer pattern but reduced VWF activity/VWF:Ag, was observed in patients with p.Arg1315Cys and p.Arg1399Cys, so they were defined as classical 2M. Given a reduction in VWF:CB/VWF:Ag ratio and a normal VWF activity/VWF:Ag ratio, both p.Ala1716Pro and p.Val1820Glu variants were classified as type 2MCB.
List of VWF variants with discrepant classification in the literature
| Variant | N of patients | VWF activity/ VWF:Ag ratio | VWF:CB/ VWF:Ag ratio | VWD diagnosis* | VWD type in literature |
|---|---|---|---|---|---|
| p.Tyr1107Cys | 1 | 0.63 | 0.9 | 2A(IIE) | 2A(IIE)8,27 or type 147 |
| p.Cys1130Phe | 2 | 0.65 (0.56-0.75) | 0.27 (0.2-0.35) | 2A(IIE) | 2A(IIE)27,46 or type 149,53 |
| p.Tyr1146Cys | 1 | 0.62 | 0.25 | 2A(IIE) | 2A(IIE)9,27,54 or type 154,46,55 |
| p.Arg924Gln-p.Arg1315Leu† | 7 | 0.45 (0.24-0.68) | 0.57 (0.38-0.95) | 2M/2A | 2A,28,54 2M,48 2A(IIE),56 or type 154 |
| p.Arg1315Cys | 3 | 0.4 (0.32-0.51) | 0.8 (0.53-0.93) | 2M | 2A,38,39,54 2M,8,27,47,54 type 18,38,47,51,54 or 2A/2M28 |
| p.Arg1374His | 24 | 0.25 (0.16-0.54) | 0.51 (0.19-0.85) | 2M/2A | 2A,28,29,53,54 2M,27,41,46,54 or 1C38 |
| p.Arg1374Cys | 6 | 0.34 (0.15-0.66) | 0.57 (0.1-0.61) | 2M/2A | 2A,38,54 2M,27,41,53,57 1C,38 or 2A/2M28 |
| p.Arg1399Cys | 3 | 0.33 (0.29-0.52) | 0.86 (0.62-1.1) | 2M | 2M8,45,46 or 2U47 |
| p.Ala1716Phe | 15 | 0.84 (0.5-1.1) | 0.63 (0.37-1) | 2M(CB) | 2M(CB)27 or type 143 |
| p.Val1820Glu | 2 | 0.8 (0.77-0.83) | 0.31 (0.3-0.33) | 2M(CB) | 2M(CB)44 or type 144 |
| Variant | N of patients | VWF activity/ VWF:Ag ratio | VWF:CB/ VWF:Ag ratio | VWD diagnosis* | VWD type in literature |
|---|---|---|---|---|---|
| p.Tyr1107Cys | 1 | 0.63 | 0.9 | 2A(IIE) | 2A(IIE)8,27 or type 147 |
| p.Cys1130Phe | 2 | 0.65 (0.56-0.75) | 0.27 (0.2-0.35) | 2A(IIE) | 2A(IIE)27,46 or type 149,53 |
| p.Tyr1146Cys | 1 | 0.62 | 0.25 | 2A(IIE) | 2A(IIE)9,27,54 or type 154,46,55 |
| p.Arg924Gln-p.Arg1315Leu† | 7 | 0.45 (0.24-0.68) | 0.57 (0.38-0.95) | 2M/2A | 2A,28,54 2M,48 2A(IIE),56 or type 154 |
| p.Arg1315Cys | 3 | 0.4 (0.32-0.51) | 0.8 (0.53-0.93) | 2M | 2A,38,39,54 2M,8,27,47,54 type 18,38,47,51,54 or 2A/2M28 |
| p.Arg1374His | 24 | 0.25 (0.16-0.54) | 0.51 (0.19-0.85) | 2M/2A | 2A,28,29,53,54 2M,27,41,46,54 or 1C38 |
| p.Arg1374Cys | 6 | 0.34 (0.15-0.66) | 0.57 (0.1-0.61) | 2M/2A | 2A,38,54 2M,27,41,53,57 1C,38 or 2A/2M28 |
| p.Arg1399Cys | 3 | 0.33 (0.29-0.52) | 0.86 (0.62-1.1) | 2M | 2M8,45,46 or 2U47 |
| p.Ala1716Phe | 15 | 0.84 (0.5-1.1) | 0.63 (0.37-1) | 2M(CB) | 2M(CB)27 or type 143 |
| p.Val1820Glu | 2 | 0.8 (0.77-0.83) | 0.31 (0.3-0.33) | 2M(CB) | 2M(CB)44 or type 144 |
If the number of patients was more than one, the median (range) of values was used.
The final diagnosis of patients was established considering both ratios of VWF activity/VWF:Ag and VWF:CB/VWF:Ag as well as the VWF multimer profile. Type 2A(IIE) showed an aberrant subband structure in multimer pattern, with a lack of the satellite bands but pronounced inner bands of the triplet. Type 2M/2A patients had decreased VWF activity/VWF:Ag and VWF:CB/VWF:Ag ratios, more pronounced in the former, and they had slight loss of large VWF multimers (supplemental Figures 1-3). Type 2M had a reduced VWF activity/VWF:Ag ratio, with a normal VWF:CB/VWF:Ag ratio; however, in type 2MCB, the opposite was observed.
All the cited references reported p.Arg1315Leu alone, except Michiels et al,56 in which p.Arg1315Leu was found in cis position with the p.Arg924Gln variant, the same as in our study.
Discussion
Type 2 VWD results from a variety of VWF qualitative defects, including multimerization defects and altered interactions with GPIb, ADAMTS-13, FVIII, or collagen.20 Generally, a battery of tests is required to establish the correct diagnosis of type 2.21 Because of the clusterization of variants in some specific functional domains of VWF, once an accurate biochemical diagnosis is obtained, the genetic diagnosis of type 2 is straightforward and confirmatory.22
This study was conducted in one of the largest cohort of patients with VWD type 2. We evaluated the laboratory phenotypic and genetic profiles of 321 patients with VWD type 2 included in the Milan cohort in the timeframe of 2 decades. Accordingly, patients were classified as type 2A (31%), 2B (26%), 2M (35%), or 2N (8%).
In general, type 2A patients showed lower laboratory results for VWF tests than other groups, except in type 2M, with the lowest medians of VWF:Ag and VWF activity (Table 1). The overall median VWF activity/VWF:Ag ratio in type 2A, 2M, and 2B was <0.7; whereas, it was normal for type 2N, 2B NY, and 2MCB. Previously, a cutoff of <0.6 was recommended to diagnose patients with type 2 (2A, 2B, and 2M),23,24 although the new guidelines recommend 0.7.25 However, we had several genetically confirmed type 2 patients (2N and 2MCB not included) with a ratio of VWF activity/VWF:Ag ≥0.6 (n = 54) or ≥0.7 (n = 34). Patients with a ratio of ≥0.7 included those with type 2A(IIE) (n = 9), classical 2B (n = 10), 2B NY (n = 11), and classical 2M (n = 4). Together, these findings suggest that not all type 2 patients necessarily show a VWF activity/VWF:Ag ratio <0.6 and that a cutoff value of <0.7 would avoid missing more type 2 patients.
In total, 82 different VWF variants were identified. VWD type 2 is primarily caused by missense variants,15,26 and this is confirmed by our results, where 78% of identified variants were missense. Gene conversions, resulting from a series of missense variants, were found in 12% of our type 2 population. Synonymous (1%) and splice (4%) variants and deletions (5%) were also observed. Except in 1 patient with type 2A(IIC) and 4 with type 2N (as a result of the recessive nature of inheritance), none of the variants identified introduced a premature termination code; rather, all of them allowed the synthesis of VWF mutants. The proportion of different subtypes of type 2 in our study (2M>2A>2B>2N) was similar to that in French cohort27 but different from those in the Spanish28 and US cohorts.29 At variance, in a study from Italy,30 among cases with type 2, type 2B was the most common subtype, followed by 2A, 2M, and 2N. We found that type 2B NY, 2A(IIE), and 2MCB were not so rare in our population. In a previous Italian investigation, the 2B NY prevalence in the type 2B population was ∼9%,31 but in the present study, it was 18% among the index cases. Given that type 2B NY usually shows laboratory results similar to those of mild type 1 or the low VWF condition, only by performing RIPA or genetic testing can they be correctly identified. In the past, we did not analyze the genotype of type 1 and low VWF patients, and RIPA testing was used only in some cases; therefore, it is likely that the prevalence of 2B NY in cases referred to our center is underestimated. A frequency of 25% for type 2A(IIE) among the index cases with type 2A was found, consistent with reports from Germany (29%)9 and France (34.5%).27 Type 2MCB, always with variants at the A3 domain, represented 20% of the type 2M index cases. However, because collagen types IV and VI were not used for the VWF:CB assay in our study, rare variants in the A1 domain (leading to reduce collagen binding) might have been missed.32,33
Thirty-one unique variants were found in all 2A subtypes. p.Ile1628Thr, p.Ser1506Leu, and p.Arg1597Gln were the most common variants in 2A(IIA), similar to the Spanish cohort for p.Ile1628Thr and p.Ser1506Leu28 and the French cohort for p.Ile1628Thr,27 but other common variants in these cohorts were not recurrent in our study. Among all type 2A in the Zimmerman cohort, substitutions of Arg1597Trp/Gln were frequent.29 In 2A(IIE), of 10 distinct variants, a synonymous variant (p.Cys1130Cys), responsible for the skipping of exon 26 (p.Pro1127_Gly1180delinsArg),34 was the most common. In contrast, in the French cohort, p.Tyr1146Cys, p.Cys1101Arg, and p.Cys1157Phe were common,27 whereas the last 2 variants were not found at all in our population. In classical 2B, 15 distinct variants were described, mostly p.Arg1308Cys, p.Arg1341Trp, and p.Val1316Met, at variance with the French and Zimmerman cohorts.27,29 In type 2B NY, 6 unique gene conversions were found. We identified 26 different variants in type 2M, including 17 in classical 2M, 4 in 2M/2A, and 5 in 2MCB. In classical 2M, p.Ala1377Val-Arg1379Cys and p.Arg1399Cys were commonly found, at variance with the other cohorts.27,29 The variants p.Ala1377Val and Arg1379Cys are located in the same allele, and their combination has so far been found only in our population.35 p.Arg1374His and p.Arg1374Cys were common among type 2M/2A cases, different from the Spanish cohort.28 p.Ala1716Pro was the most common variant in 2MCB patients, distinct from the common variants in the French (p.Leu1696Arg)27 and the Spanish cohorts (p.Arg1399His),28 which were not found in our population. In contrast to the other cohorts, where several type 2N variants were identified, we found only p.Arg854Gln.
In type 2A, 49% of the variants occurred in the A2 domain, leading to type 2A(IIA) with an enhanced susceptibility to ADAMTS-13 activity. Whereas, 32% were found at the D3 domain consisting of 2A(IIE) and only 3% associated with 2A(IIA) which were found at the A1 domain. Two variants (6%) in compound heterozygosity were found at the D1-D2 domains, causing 2A(IIC). Type 2A(IIH), characterized by the loss of the largest plasma and platelet multimers and absence of the triplet structure, was found in a patient with 3 variants (10%). In this case, a defective multimerization process for p.Arg202Trp and p.Cys849Tyr and intracellular survival for p.Cys849Tyr were already established.36 In type 2B, 67% of variants were found at the A1 domain, and the rest (33%) were located at the D3-A1 junction, described as 2B NY. For type 2M, 80% of variants were at the A1 domain (classical 2M and 2M/2A), whereas 20% were at the A3 domain (2MCB). In type 2N, all patients had the p.Arg854Gln (D′ domain) variant, although compound heterozygosity with type 1 and type 337 variants was also found.
Several variants are reported in the literature to be associated with different VWD types, and the classification of these variants is still a matter of debate. In a recent report from the Zimmerman cohort,38 p.Arg1374Cys was associated with type 2A and type 1C and p.Arg1374His with type 1C. p.Arg1315Leu/Cys substitutions are other variants of controversial diagnoses.39-42 p.Arg1374Cys and p.Arg1315Cys were classified as type 2A/2M in the Spanish study,28 but p.Arg1315Cys was reported as type 2A or 1C in the Zimmerman cohort.38 We classified p.Arg1315Leu and p.Arg1374His/Cys as 2M/2A, because patients had markedly decreased VWF activity/VWF:Ag ratios but only mildly decreased VWF:CB/VWF:Ag ratios along with a modest loss of large VWF multimers (Table 4). The reduced ability of mutated A1 domains to bind GPIb is distinctive of type 2M, whereas the loss of high molecular weight multimers and decrease in VWF:CB are distinctive of type 2A (supplemental Figures 1-3). Variants p.Ala1716Pro and p.Val1820Glu (A3 domain) have been associated with type 1 or 2MCB.43-45 We categorized both variants as 2MCB, because they presented abnormal VWF:CB/VWF:Ag ratios (Table 4). p.Arg1399Cys is reported as type 2M or 2U or type 1,8,46,47 and we classified this variant and p.Arg1315Cys as classical 2M (Table 4), considering the normal VWF:CB/VWF:Ag ratios and multimeric pattern contrasting with a decreased activity/VWF:Ag ratio. Three variants in the D3 domain (p.Tyr1107Cys, p.Cys1130Phe, and p.Tyr1146Cys) have been associated with type 1 and type 2A.48-51 Carriers of these variants showed an aberrant subband multimer pattern with a lack of satellite bands but a pronounced inner band of the triplet structure. Therefore, they were defined as type 2A(IIE) in our and the French studies.27
Of 82 variants identified, 8 were novel variants (Table 3). p.Leu893Gln (in compound with p.Arg854Gln) was predicted to be pathogenic. The patient with this variant showed reduced VWF and FVIII levels. Variants p.Cys1126Tyr, p.Cys1142Phe, and p.Leu1281Arg were identified in type 2A(IIE) and were defined as pathogenic variants by SIFT, Polyphen, CADD, PROVEAN, and M-CAP. In patients with classical 2M or 2MCB, we found 2 novel variants (p.Arg1426Pro and p.Ser1731Leu). p.Arg1426Pro was determined to be a pathogenic variant by all in silico tools, and the patient showed a ratio of 0.31 for VWF activity/VWF:Ag but a normal VWF:CB/VWF:Ag ratio (0.79), with a normal multimer pattern. p.Ser1731Leu resulted in severely reduced collagen binding but normal VWF values (Table 3). All algorithms predicted it as a damaging variant. p.Leu1657Pro was predicted as damaging, and the 2A(IIA) patient showed a loss of large multimers with a very low VWF activity/VWF:Ag ratio (0.22).
A limitation of our study is that of using the target sequencing approach. However, because of the negative-dominant nature of most VWD type 2 variants, the possibility of other existing variants is rare, although not completely excluded. In addition, almost all the identified variants are well known to be responsible for VWD type 2. Nowadays next-generation sequencing makes the genetic analysis more straightforward, even though in developing countries our approach is still useful. Another limitation of the study is that the bleeding scores of the patients were not available.
In conclusion, given the complexity of VWD type 2 diagnosis and the necessity of performing several phenotypic tests, genetic analysis of VWF in type 2 patients is beneficial and sometimes crucial to establish the correct diagnosis. The results of our large cohort study show that VWD type 2 is invariably due to the variants that do not prevent the synthesis of the protein, and 88% of patients have missense variants.
Acknowledgments
The authors acknowledge P.M. Mannucci for critical advice and L.F. Ghilardini for illustration work.
This work was partially supported by the Italian Ministry of Health-Bando Ricerca Corrente.
Authorship
Contribution: O.S., L.B., and F.P. conceived and designed the study; O.S. collected and analyzed data and wrote the manuscript; L.B. wrote the manuscript; M.T.P. and A.C. performed the genetic tests; P.C. and G.C. performed the phenotypic tests; S.M.S. and E.B. clinically evaluated patients; L.B. and F.P. critically revised the manuscript; and all authors approved the final manuscript.
Conflict-of-interest disclosure: F.P. reports participation in educational meetings and advisory boards of Sanofi, Sobi, Takeda, Roche, and Biomarin. The remaining authors declare no competing financial interests.
Correspondence: Flora Peyvandi, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Angelo Bianchi Bonomi Hemophilia and Thrombosis Center, Milan, Italy; and Department of Pathophysiology and Transplantation, Università degli Studi di Milano, Via Pace 9, 20122 Milan, Italy; e-mail: flora.peyvandi@unimi.it.

