


Abstract
The presence of blood flow influences the interaction between von Willebrand factor (VWF) and blood cells, affecting characteristics of forming blood clots. The interactions between coagulation and inflammation have mainly been studied in thrombosis models, but it remains unclear whether these interactions might also play a role in reduced bleeding in patients with bleeding disorders. In this systematic review, we provide an overview of the literature investigating the interactions between VWF and blood cells in flow models. For article selection, a systematic search was performed in Embase, Medline-Ovid, Cochrane Library, Web of Science databases, and Google Scholar. After selection, 24 articles were included. These articles describe direct or platelet-dependent interactions between VWF and neutrophils, monocytes, erythrocytes, or lymphocytes under different flow conditions. Almost all the described interactions required the presence of activated platelets. Only erythrocytes, monocytes, and natural killer cells were capable of directly binding the VWF multimers. Overall, interactions between VWF and blood cells mainly occurred in the presence of platelets. Because of the large variation in study design and used flow rates, further research is necessary to compare the results between studies and draw firm conclusions on when and under what conditions these interactions can occur. After our findings, many questions remained unanswered. This review might provide a starting point for future research. Extended knowledge on the influence of blood flow on VWF and blood cell interactions can contribute to improved understanding of the variation in bleeding in patients with bleeding disorders.
Introduction
Von Willebrand factor (VWF) is a multimeric glycoprotein that is synthesized in endothelial cells and megakaryocytes. VWF plays a major role in primary hemostasis, where it mediates platelet adhesion and platelet-platelet interactions to form an initial clot and consequently prevent blood loss at the site of injury. Defects in the VWF protein can result in von Willebrand disease (VWD), which is the most common inherited bleeding disorder affecting 1 in 1000 to 10 000 individuals.1,2 VWD can be divided into 3 different types: a partial quantitative deficiency of VWF (type 1), a qualitative disorder in the VWF protein (type 2), or a complete quantitative deficiency of VWF (type 3).3 The diagnosis of VWD is based on 3 different aspects: results from laboratory testing, assessment of the bleeding history of the patient, and assessment of the family’s bleeding history.4
The bleeding assessment tool, based on the patient’s bleeding history, frequently reflects the severity of disease in patients with VWD.5 However, in some patients, the bleeding score is not the best tool with which to assess disease severity. The bleeding score is highly influenced by the number of bleeding challenges and the age of the patient, which might lead to an underestimation of the bleeding risk in both children and adults.6-8 In addition, bleeding is not always related to a bleeding disorder; 20% of the healthy population has also experienced an increased number of bleeding events without an identifiable bleeding disorder.9,10 Genotyping has been suggested as an alternative tool to diagnose patients with VWD. However, no genetic mutation can be identified in 45% to 56% of patients with confirmed type 1 VWD that results in reduced VWF levels.8,11 These patients might have mutations outside the VWF gene that alters its functioning. The application of flow models can provide more information about the malfunctioning of VWF in these patients.
When only looking at the VWF genotype of a patient, environmental influences and condition-specific cell-cell interactions during coagulation that contribute to the clotting capacity are missed. As an example, different functions of the endothelium are affected by changes in flow, such as permeability of the endothelial layer, proliferation, apoptosis, coagulation response, and vascular remodeling.12,13 VWF also plays a role in the maintenance of coagulation factor VIII levels. Binding of VWF to coagulation factor VIII prevents rapid degradation of factor VIII.14 Consequently, VWF levels also directly influence coagulation factor activation and fibrin formation. Finally, VWF can link coagulation and inflammation. Previous studies performed in mice have reported that binding of neutrophils to VWF resulted in neutrophil extracellular trap (NET) formation and an increased incidence of deep vein thrombosis.15,16
The interactions between coagulation and inflammation have mainly been studied in light of thrombosis, but it remains unclear whether these interactions might also play a role in bleeding in patients with a bleeding disorder. The role of VWF in both coagulation and inflammation remained inconclusive based on studies in VWF-deficient pig models.17 The introduction of different flow models has provided a new approach with which to study the interactions between VWF and blood cells, which include neutrophils, monocytes, erythrocytes, and lymphocytes. In this systematic review, we will provide an overview of the studies that describe the interactions between VWF and blood cells and blood cell components, whether platelet dependent or independent. Only studies that made use of a flow model setup were included in this systematic review. The interactions that are described in this review are summarized in the figure of the visual abstract. Although no clear evidence was found in these studies, we hypothesize that interactions between platelets, other blood cells, and VWF might be a contributing factor to the heterogenicity in bleeding phenotypes of patients with the same congenital bleeding disorder of primary hemostasis. The interactions between VWF, platelets, and other blood cells identified in this review could function as a starting point for future research. This research will provide more insights into the impact of interactions between VWF and inflammatory cells, erythrocytes, and coagulation on the bleeding phenotype.
Interactions under flow
Interactions between VWF, platelets, and blood cells
Hemostasis is the process where a hemostatic plug is formed in order to prevent bleeding upon damage of the vessel. Upon injury, smooth muscle cells in the vessel wall will contract, a process called vasoconstriction, which increases shear stress. Binding of platelets to exposed proteins in the extracellular matrix, such as collagen (integrin α1β2 and glycoprotein VI), fibronectin (integrins αIIbβ3, α5β1, and αvβ3), and thrombospondin (integrins αvβ3 and αIIbβ3 and CD36), leads to platelet activation.18-20 Furthermore, platelet activation can also be triggered by soluble agonists, such as endothelial-derived ADP, thromboxane A2 (released from other activated platelets), and thrombin. Simultaneously with platelet activation, VWF is released from the endothelial Weibel-Palade bodies into the bloodstream. Released VWF multimers uncoil in the presence of high shear stress, exposing the different VWF domains. The glycoprotein Ibα, part of the glycoprotein Ibα-IX-V complex on platelets, interacts with the A1 domain of VWF, which results in additional platelet binding and platelet activation.21,22 Upon platelet activation, receptors such as αIIbβ3 integrins, CD40L (CD154), and P-selectin become highly expressed on the cell surface. These receptors are important mediators in interactions with fibrinogen and other circulating cells in the blood.
Blood flow and clot composition
Shear stress, as a result of the presence of blood flow, is important not only in the uncoiling of VWF multimers but also as a contributor to clot composition and clot characteristics. As an example, the difference in clot composition in thrombi formed under low and high blood flow can be observed in thrombi obtained from patients with venous or arterial thrombosis. Arterial thrombi, formed under high blood flow and consequently high shear stress, are mainly composed of fibrin and activated platelets.23,24 Other blood cells are less abundant. In contrast, venous thrombi, formed under low blood flow and low shear stress, mainly contain large numbers of erythrocytes, which are trapped in web-like structures that consist of fibrin and activated platelets.23,24
Inflammation and clot composition
Inflammation can also have an effect on clot composition. Alterations in clot composition resulting from the presence of immune cells within the thrombus influence properties of the thrombus.25 The processes of inflammation and coagulation are tightly linked in order to prevent the spreading of pathogens upon trauma. Previous studies have described interactions between activated platelets, the endothelium, and leukocytes, which can contribute to inflammation and thrombosis.16,26,27 For example, there is cross-talk between the activation of the intrinsic coagulation pathway and the activation of the complement system.28 Also, VWF and activated platelets play an important role in the recruitment of immune cells to the site of an injury. The required interactions for this recruitment will be discussed in this review. In contrast, in a study by Henn et al,29 CD40L, released from platelets, was reported to temper the activation of the immune system.
Clinical relevance
It is important to study the variety in clot composition. The presence of different blood cells during thrombus formation can influence the coagulation cascade. Consequently, the properties of a thrombus might be changed, which affects other processes such as fibrinolysis. One of the examples that illustrates this is that monocytes, activated via P-selectin, start to express tissue factor, which contributes to increased cleavage of factor VII and more rapid fibrin formation.26 One could imagine that inflammation might push the balance of hemostasis toward thrombosis in healthy individuals. In contrast, in patients with a bleeding disorder, the presence of these immune cells during thrombus formation might also affect the bleeding phenotype. With more knowledge of the influence of blood flow on the interaction of VWF with blood cells, and ultimately the clot composition, we might be able to explain some of the variation in bleeding phenotype and treatment response observed in patients with VWD.
Methods
This systematic review was performed according to the Preferred Reporting Items for Systematic Reviews and Analyses guidelines.30 The flow scheme for article selection according to this method is presented in Figure 1. The full search strategy and a table with the experimental setup per study are included in the data supplement.
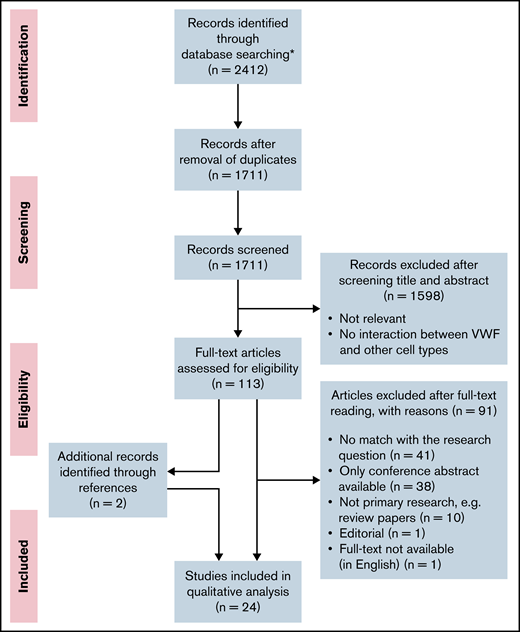
PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) flow diagram. Flow diagram presenting the article search, screening, and study selection. *No additional records were identified through other sources.
PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) flow diagram. Flow diagram presenting the article search, screening, and study selection. *No additional records were identified through other sources.
Article search
The first search was completed on 29 June 2020 in 5 different databases: Embase, Medline-Ovid, Cochrane Library, Web of Science databases, and Google Scholar. The search was repeated on 22 February 2021. The search strategy can be found in the data supplement. No limit was set on year of publication.
Study selection
After removal of duplications, the search resulted in 1711 articles. All articles were screened independently by 2 reviewers (R.A.A. and J.J.d.V.). The first selection of relevant articles was made on the basis of information in the title and abstract. In the second selection round, 113 articles were screened on relevance on the basis of the full text. Articles that did not match the research question, as well as conference abstracts, reviews, and articles that were not available in full text or in English, were excluded. The number of articles excluded and the reasons for exclusion are listed in Figure 1. Additional relevant articles identified while reading the full-text articles were also assessed for inclusion and included if there was a match with the research question. During the study selection, 2 additional articles were found while reading the full-text articles, which were also included in the review. These 2 articles were not found in our search because the keywords as flow, shear stress, or microfluidics were missing in the title and abstract. In total, we included 24 studies. In case of disagreement, consensus about the articles was reached through discussion.
Data extraction
To prevent bias, data were independently extracted from the articles by 2 researchers (R.A.A. and J.J.d.V.). The following data were collected: first author, publication year, method of experiments (eg, type of flow model, use of different coatings, use of plasma with the addition of blood cells or whole blood, and applied flow rates), and type of interaction and (potential) mechanism of action.
Results
Study characteristics
A large variation in study design between the included articles was present, including type of flow chamber, type of coating of the channel, variation in flow rate, and method of analysis. A short overview of the differences in study design will be given in this section. An overview of the experimental setup of each study is included in the data supplement (supplemental Table 1).
First, different types of flow devices were used among the different studies. A parallel flow chamber was the most common flow device. Examples of such parallel flow chambers are the VenaFluoro8+ microchip (Cellix), the BioFlux (Fluxion), and the Luer microslide (Ibidi).
To study the interaction with VWF, the flow device channel was either coated with VWF protein or lined with endothelial cells. To release VWF from the endothelial cells, cells were activated using a reagent such as histamine.
Finally, either plasma, with or without the addition of platelets and other blood cell types, or whole blood was used to flow through the channel to study the interactions of each cell type with VWF. Studies used different shear rates (50-10 000/s) and shear stresses (0.5-100 dyn/cm2). In this review, we made a distinction between low (venous) flow rates, resulting in low shear rates (50-100/s) and shear stress (0.5-3.0 dyn/cm2), and high (arterial) flow rates, resulting in high shear rates (500-10 000/s) and shear stress (40-100 dyn/cm2). The interactions of VWF with 6 different blood cell types are described in this systematic review. The interactions were studied using retention assays, inhibitory antibodies, and fluorescence microscopy.
Interaction between VWF and erythrocytes
The number of erythrocytes is much lower in arterial thrombi, formed under high flow rates, than in venous thrombi.31,32 We did not find studies describing interactions between VWF and erythrocytes under high shear stress. However, multiple studies showed that the hematocrit is highly important for the binding of platelets to VWF.33-35 VWF-bound platelets are important mediators for the interaction of other blood cells with VWF. The presence of erythrocytes alters the viscosity of the blood, which results in the promotion of platelet margination supporting platelet adhesion to the vessel wall.35 In addition, rotation of erythrocytes contributes to enhanced diffusion of platelets within the blood vessel.36-38 Under lower shear stress in small vessels, erythrocytes stack together, which further enhances platelet deposition.39 A detailed discussion of the effect of hematocrit on platelet binding to VWF is beyond the scope of this review.
Venous thrombi, in contrast, are characterized by a strong presence of erythrocytes. Multiple studies showed that erythrocytes can roll on and adhere to ultralarge VWF multimer (ULVWF) strands released from activated endothelial cells under low shear stress up to 1 dyn/cm2.40-44 Results from a study by Smeets et al42 demonstrated that part of this observed adhesion could be attributed to intracellular calcium-loaded erythrocytes. Intracellular calcium-loaded erythrocytes are able to bind ULVWF independently of platelets. The mechanism of action for this binding was not identified, but the adhesion of erythrocytes to VWF was not affected by the addition of platelets. This result suggests that erythrocytes and platelets have a different binding domain within VWF, which indicates that intracellular calcium-loaded erythrocytes do not adhere to the A1 domain.42 Erythrocytes can also bind to intermediate-weight VWF, facilitated by activated platelets, once the wall shear stress is reduced. A reduction in wall shear stress can, for example, occur upon fibrin network formation.43
Even though several studies described interactions between erythrocytes and VWF under low shear stress conditions, this interaction was often observed for only small numbers of erythrocytes. However, the adhesion of erythrocytes is much more enhanced in patients with sickle cell (SS) disease. Multiple studies showed that under low shear stress, increased adherence of SS erythrocytes to VWF was observed compared with adhesion of regular erythrocytes to VWF as described in the previous section.44-46 Increased adhesion of SS erythrocytes is induced via the expression of annexin V on their cell membrane. Annexin V is then able to directly interact with the A1 domain of uncoiled VWF.47 In addition, adhesion of SS erythrocytes to activated platelets is further promoted in plasma that contained a soluble form of VWF, in which the A1 domain is not exposed.48 This increased adhesion of SS erythrocytes in the presence of soluble VWF suggests that SS erythrocytes can bind to other domains of VWF apart from the A1 domain. Finally, the study by Walmet et al49 suggests that integrins on the membrane of SS erythrocytes are involved in binding to ULVWF, but the binding place within the VWF protein remains unclear.
To conclude, interactions between VWF and normal erythrocytes, direct or platelet dependent, were only described to occur under low shear flow. All interactions between VWF, platelets, and erythrocytes are depicted in Figure 2.
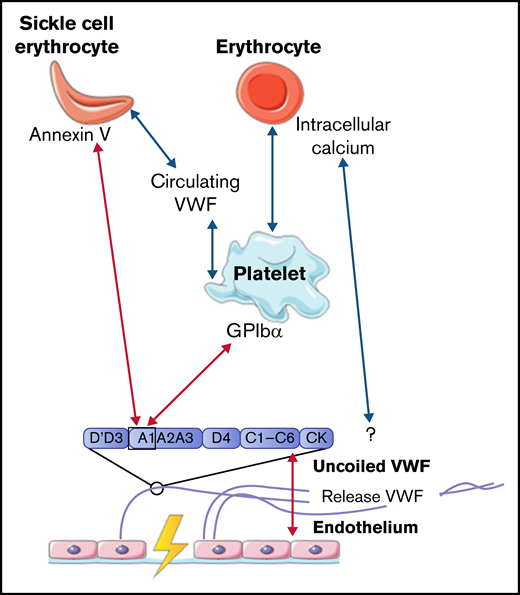
Interactions between VWF and erythrocytes studied in flow models. Identified interactions between VWF multimers, platelets, and (SS) erythrocytes under high shear flow (red arrows) and low shear flow (blue arrows). GP, glycoprotein.
Interactions between VWF and erythrocytes studied in flow models. Identified interactions between VWF multimers, platelets, and (SS) erythrocytes under high shear flow (red arrows) and low shear flow (blue arrows). GP, glycoprotein.
Interaction between VWF and neutrophils
Upon inflammation, neutrophils need to transmigrate through the vessel wall to the site of infection; this is done via diapedesis. As a first step, neutrophils roll over the vessel wall to reduce speed, before full retention is reached. Multiple studies observed that the rolling and retention of neutrophils were facilitated by platelets adherent to released VWF.50-54 To show the dependence on platelets and VWF for adhesion, flow devices were directly coated with either VWF or human umbilical vein endothelial cells that were stimulated with histamine to induce VWF release. VWF strands were decorated with platelets, before flowing with neutrophils containing plasma or whole blood. Although the presence of VWF is required to initiate the rolling process, no direct interactions between neutrophils and VWF were found.50 -52,55 -58 Platelet binding to VWF and activation of platelets initiate expression of various receptors on the cell surface, including P-selectin.50 Under high shear stress up to 20 and 40 dyn/cm2, neutrophils start to roll over ULVWF, released by activated endothelial cells during inflammation.50 This process is initiated by the temporary binding of neutrophils to high-density P-selectin on VWF-bound platelets. Neutrophils continue to roll until their speed has decreased and total adhesion is reached. Activation of platelets also induces the expression of αIIbβ3 on the surface. CD16+ neutrophils or neutrophils activated with lipopolysaccharide or tissue necrosis factor α can bind to this receptor via the expression of the SLC44A2 receptor and human neutrophil antigen 3a under low shear conditions.51,52
The mechanical stress that is created upon the binding of neutrophils to platelets induces the formation of NETs.51 NET-derived double-strand DNA can directly bind to the A1 domain of VWF in a charge-dependent manner.55,57 The release of other neutrophil content during NET formation can also influence the interaction between VWF and platelets. For example, enzymes such as matrix metalloproteinase-8, matrix metalloproteinase-9, and neutrophil elastase can enhance platelet binding under high flow conditions, whereas other proteins, like human neutrophil peptide 1, prevent platelet binding.56,58
In conclusion, all identified interactions between VWF and neutrophils were facilitated by activated platelets. Only NET-derived double-strand DNA can bind directly to the A1 domain of VWF. All interactions between VWF, platelets, and neutrophils are depicted in Figure 3.

Interactions between VWF and neutrophils studied in flow models. Identified interactions between the A1 domain of VWF multimers, platelets, neutrophils, and NETs under high shear flow (red arrows) and low shear flow (blue arrows). ds, double strand; GP, glycoprotein.
Interactions between VWF and neutrophils studied in flow models. Identified interactions between the A1 domain of VWF multimers, platelets, neutrophils, and NETs under high shear flow (red arrows) and low shear flow (blue arrows). ds, double strand; GP, glycoprotein.
Interaction between VWF and monocytes
In concordance with neutrophils, monocytes also interact with VWF in a 2-step process. The first step is the rolling of monocytes over VWF strands to reduce speed. Monocytes mainly roll on VWF strings that are rich in platelets. This interaction is mediated via the binding of P-selectin glycoprotein ligand-1 (PSGL-1) on the cell membrane of monocytes to high-density P-selectin that is expressed on activated platelets (Figure 4).54,59 In the second step, monocytes firmly adhere to the platelets on VWF strands to reach full retention.50 This process is observed both under low and high shear conditions.50,54 Then, monocytes migrate in the direction of the endothelial cells, where there is less platelet adhesion.50,54 Under low shear flow conditions, CD40 ligand (soluble CD154), released from platelets, can also initiate and further enhance the release of ULVWF from endothelial cells, which results in increased platelet deposition and increased platelet-monocyte interactions.54,59,60 Monocytes can also bind directly to VWF in a similar 2-step process under low shear stress. First, short adhesion was observed via PSGL-1 and the A1 domain on VWF.53 Then, firm adhesion was established via the interaction of β2-integrins on monocytes with the A1-A2-A3 domain and the dimeric D′-D3 domain of VWF.53
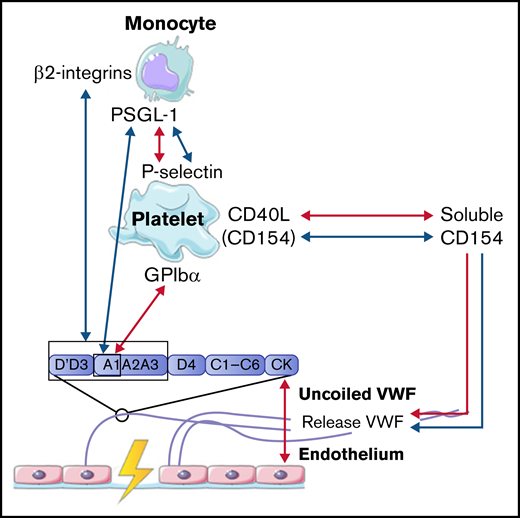
Interactions between VWF and monocytes studied in flow models. Identified interactions between the A1 domain of VWF multimers, platelets, and monocytes under high shear flow (red arrows) and low shear flow (blue arrows). GP, glycoprotein.
Interactions between VWF and monocytes studied in flow models. Identified interactions between the A1 domain of VWF multimers, platelets, and monocytes under high shear flow (red arrows) and low shear flow (blue arrows). GP, glycoprotein.
Taken together, these results indicate that the interaction between VWF and monocytes is dependent on activated platelets, like neutrophils, or can occur directly via PSGL-1 and β2-integrins on monocytes. The interactions between VWF, platelets, and monocytes are depicted in Figure 4.
Interaction between VWF and lymphocytes
Lymphocytes, including T cells, B cells, and natural killer cells, were described to interact with activated platelets on VWF. T cells are able to interact with platelet receptors P-selectin and αIIbβ3.51,61 B cells only adhere through P-selectin.51,61 Natural killer cells were shown to adhere to VWF-coated surfaces under flow, most likely in a platelet-dependent way, but the specific receptors involved in this interaction were not identified in this study.61 The identified interactions between VWF, platelets, B cells, T cells, and natural killer cells are depicted in Figure 5.
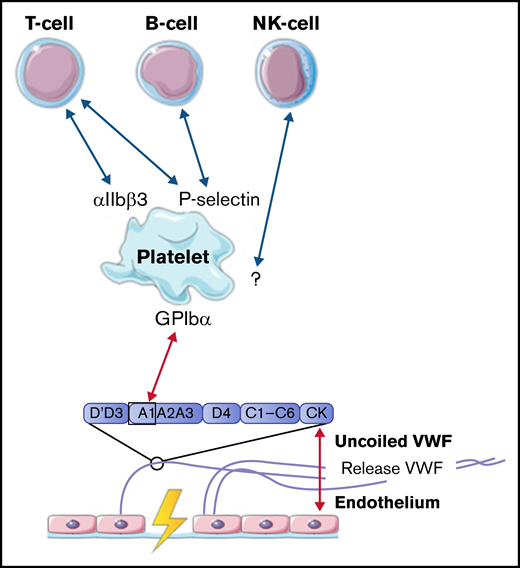
Interactions between VWF and lymphocytes studied in flow models. Identified interactions between the A1 domain of VWF multimers, platelets, B cells, T cells, and natural killer (NK) cells under high shear flow (red arrows) and low shear flow (blue arrows). GP, glycoprotein.
Interactions between VWF and lymphocytes studied in flow models. Identified interactions between the A1 domain of VWF multimers, platelets, B cells, T cells, and natural killer (NK) cells under high shear flow (red arrows) and low shear flow (blue arrows). GP, glycoprotein.
Discussion
In this systematic review, we present an overview of the known interactions between VWF and blood cells under flow in vitro, based on 24 articles. An overview of all identified interactions are depicted in the visual abstract. A majority of the interactions between blood cells and VWF require the exposure of the A1 domain under high shear conditions. Blood cells can bind this A1 domain either directly or indirectly, via platelets that bind to this domain. Almost all interactions described in this systematic review between blood cells and VWF are dependent on platelets, and the interaction between platelets and blood cells takes place only under lower shear flow. In contrast, the interaction between the A1 domain in VWF and platelets depends on the uncoiling of VWF multimers, which requires high shear flow. The authors of each study do not address any potential mechanism that can explain the change in flow that is necessary for A1 domain exposure. One of our suggestions is that the change in arterial pressure might provide a fluctuation in shear stress, resulting in low shear stress enabling platelet binding to blood cells, as well as high shear stress enabling platelet binding to VWF. A different suggestion might be that formed clots alter the flow near the vessel wall, resulting in locally reduced shear stress. This reduced shear stress then enables binding of blood cells to platelets.
As mentioned previously, the presence of erythrocytes in a clot is known to be a characteristic of a clot formed under venous shear conditions. Here, we describe several studies that showed various type of interactions between VWF and erythrocytes, direct or indirect via platelets, under low shear stress. However, these interactions are only found for small subgroups of erythrocytes, and they do not explain the high number of erythrocytes present in venous thrombi. This indicates that other mechanisms contribute to erythrocyte retention apart from the interaction with VWF. One of the explanations could be that the formation of the fibrin network and the cross-linking of fibrin fibers by factor XIII result in a decrease in flow and that erythrocytes either get trapped in the web-like structure or directly interact with the fibrin network.62-64 In addition, multiple VWF strands can also intertwine and form larger bundles, especially in the curves of a vessel. This process alters the flow, resulting in retention of erythrocytes.65
The studies in this review did not show any interaction between VWF and erythrocytes under high shear stress. A possible explanation for this observation is that the rheology of erythrocytes in blood can, depending on the diameter of the vessel, the shear stress, and the hematocrit, result in an erythrocyte cell–free layer at the site of the vessel wall, preventing direct interaction between VWF and blood cells.33,34,66 -68 However, erythrocytes are often not completely absent in arterial thrombi, which might indicate that the difference in erythrocyte counts between thrombi can still influence the clot properties. In comparison with regular erythrocytes, SS erythrocytes can directly interact with VWF or the endothelium under both high and low flow conditions. These interactions might contribute to the increased risk of thrombosis seen in patients with SS disease.69
No direct interaction between neutrophils and VWF under flow was described in any of the reported studies. Instead, all interactions were facilitated by activated platelets bound to released VWF multimers. An important process that links the coagulation and the immune response is NET formation. NETs enhance the recruitment of platelets to the site of injury and simultaneously activate the complement system. Complement activation results in activation of the contact pathway via factor XII and factor XI and in increased recruitment of platelets, further attracting more neutrophils and other immune cells.70-72
Upon NET formation, NET-derived DNA can directly interact with the A1 domain of VWF.51 Release of NETs by neutrophils can be stimulated by different processes. NET formation can be triggered by mechanical stress, as a consequence of shear stress in flow conditions. Platelets activated via toll-like receptor 4 by lipopolysaccharides on bacteria are also able to induce NET formation under very low flow conditions, where mechanical stress is almost absent.73 Furthermore, proteins released by neutrophils during NET formation also alter the binding of platelets, which further enhances binding of neutrophils and other immune cells.
Monocytes also display platelet-dependent interactions with VWF, similar to neutrophils. They use PSGL-1 to roll over VWF-bound platelets to reduce speed, enabling full retention. Stimulation of endothelial cells with platelet-derived CD154 can further enhance VWF release and monocyte retention. Additionally, monocytes could also directly interact with VWF via PSGL-1 and β2-integrin. This interaction was previously identified in experiments under static conditions.74 Results showed that the Leu-Leu-Gly sequence of β2-integrin could bind to the D3 domain as well as the A1-A2 domain of the VWF protein. Removal of the sequence, however, did not prevent the binding of monocytes, suggesting that other interactions between monocytes and the endothelium occur simultaneously.53 Apart from the interactions between monocytes, platelets, and VWF, the authors of the included studies did not further elaborate on the effect of the presence of monocytes on coagulation.
In the final part of this review, the interaction of lymphocytes with VWF was studied. Results from these studies implicated the presence of platelet-dependent interaction of VWF with T and B lymphocytes. The interaction between VWF and natural killer cells was also platelet dependent, but the receptors involved in these interactions were not identified. It should be noted that studies investigating the interaction between VWF and lymphocytes under shear stress are limited. More research is necessary to validate these interactions in order to establish the receptors involved in the interaction between these cell types, platelets, and VWF.
Overall, almost all interactions between blood cells and VWF required platelets. Platelets have been described in previous studies as important orchestrators of the thrombus architecture.75,76 After binding of fibrin fibers, platelets induce clot contraction, making the thrombus more compact. As a result, the core of the thrombus consists of dense fibrin fibers, bound by activated platelets that are mainly P-selectin+. The outer shell of the thrombus contains platelets that are less activated and lack P-selectin expression. The P-selectin+ platelets serve as a guide for leukocyte trafficking to the site of injury.75
Besides the presence of activated platelets, one could imagine that trapped cells hamper clot contraction, making the thrombus less dense.77-79 For erythrocytes, it is known that clot structure is changed in terms of stability, embolization, and susceptibility to clot lysis.80 Incorporation of erythrocytes results in a larger thrombus that is less permeable and displays increased resistance to lysis, even though the pores between fibrin fibers are much larger.77,78,81,82 It is plausible that the presence of other cells within the thrombus will have similar effects, which might contribute to increased risk of thrombosis. However, a study by Levin et al83 showed that erythrocyte-derived microvesicles that carry plasminogen on their surface can contribute to improved fibrinolysis in vitro. Further research must provide more insight into the regulatory effects of blood cells within the thrombus.
One important question is how immune cells, apart from their role in thrombus properties, influence the pathophysiological consequences of coagulation. The studies in this review as well as others observed that platelets form the link between coagulation and inflammation. Platelet activation can mediate the attraction of leukocytes to the site of injury. Neutrophils and monocytes carry tissue factor on their cell membranes and release microvesicles that are enriched in tissue factor, leading to increased coagulation.84-87 Consequently, the procoagulant activity of immune cells is often linked to thrombosis and sepsis.
In contrast, the presence of cells during coagulation might also contribute to the differences in bleeding phenotype in patients with a bleeding disorder. As stated in the Introduction, the interaction of VWF and blood cells under flow might explain some of the variation in bleeding phenotype in patients with an already established hemostasis disorder, such as VWD, as well as in a significant number of patients with an increased bleeding tendency without a clearly defined diagnosis (ie, patients with bleeding of unknown cause). Results in previous sections showed that exposition of the A1 domain within the VWF multimer is essential for platelet retention and activation, as well as direct binding of blood cells. The presence of blood cells near the site of injury can contribute to a procoagulant environment. Multiple studies showed that activated monocytes can express tissue factor.88-91 In addition, they can also produce tissue factor–loaded microparticles to transfer tissue factor to nearby platelets and neutrophils, which are not able to produce tissue factor themselves.92-97 Another example that blood cells can contribute to a procoagulant environment could be observed for erythrocytes. Studies showed that not only platelets but also erythrocytes are able to expose phosphatidylserine on their membranes.98 Phosphatidylserine plays an important role in the assembly of coagulation complexes, such as prothrombinase. These examples suggest that decreased interactions and therefore a lower presence of blood cells within the thrombus may contribute to a more severe bleeding phenotype in patients with VWD. Furthermore, the blood group expressed on erythrocytes is also linked to VWD severity, further supporting the idea that erythrocytes might influence the bleeding phenotype.99,100
In patients with hemophilia, the intrinsic pathway of coagulation is less functional, resulting in less fibrin formation. The formation of a fibrin network is important to reduce the shear flow near the site of an injury, which enables other cells to bind to VWF or activated platelets. In order to initiate fibrin formation, activation of the secondary hemostasis is required. Nonfunctional or reduced levels of coagulation factors can hamper fibrin network production, as seen in patients with hemophilia A or B. Flow studies with impaired factor VIII activity under high, intermediate, and low flow rates observed decreased platelet retention and decreased fibrin deposition.101,102 This may result in fewer interactions between VWF and blood cells and may contribute to a more severe phenotype. Furthermore, as discussed as in previous sections in this review, the presence of blood cells within thrombi can also alter the coagulation and fibrinolysis processes. These alterations might partly explain the heterogeneity in bleeding phenotype in patients with comparable low factor VIII levels.
Overall, most studies in this review described platelet-dependent interactions between VWF and blood cells. One limitation of this review is that it is difficult to compare the results between the different studies because the experimental setup, such as the type of flow chamber and the applied shear stress, varies from each study to the next (supplemental Table 1). Because many studies started with high shear stress to uncoil VWF multimers and then changed to lower shear stress to study cell adhesion, translation of the results to an in vivo setting is also complicated. Another limitation of this review is that studies that investigated the interactions in animal models were excluded from the search in order to keep the overview clear. The effect and interactions of VWF during coagulation and inflammation studied in mice models have been reviewed previously.103 Most animal studies are performed in the context of thrombosis and atherosclerosis. Only 2 mice studies addressed the impact of VWD on inflammation.104,105 Mice with severe VWD showed impaired leukocyte rolling after endothelial activation. Further analysis revealed that these mice showed impaired P-selectin expression on the endothelial surface because of insufficient Weibel-Palade body formation and secretion. A similar reduction in leukocyte rolling was observed when the same experiment was repeated after administration of anti-VWF antibodies to wild-type mice. These data further highlight the role of coagulation in inflammation. This type of phenomenon might fulfill a critical function in coagulation in patients with bleeding disorders. The lack of data on the combined role of inflammation and coagulation in hemostasis further underlines the need for more extensive research on this topic. The last limitation of this study that we would like to address is that the number of studies per cell type was limited.
Further research is necessary to establish more knowledge on the influence of blood flow on the interaction between VWF, platelets, and blood cells with the use of an experimental setup that mimics the in vivo situation. Based on the results described in this review, there are still many questions that remain unanswered. For example, most studies solely examined the interactions of 1 specific cell type, and it remains unclear how different cell types influence the functioning of one another and in what timeframe each of the interactions occur. Furthermore, there has been almost no research performed on the analysis of blood cell interaction under flow conditions and the corresponding effect on bleeding Finally, the effect of the recruitment of cells on clot formation was also undiscussed in almost all reports. The information described in this review might provide a starting point for new research in order to answer these questions. This knowledge will give more insight into the effect of blood flow on clot composition and, as a result, the potential change in clot properties, which might be an important factor in understanding the heterogeneity in bleeding phenotype in patients with an already established diagnosis or even in patients without a hemostasis diagnosis.
Acknowledgments
The authors thank M. F. M. Engel from the Erasmus MC Medical Library for developing and updating the search strategies.
This research received funding from the Netherlands Organization for Scientific Research in the framework of the National Science Agenda Programme Research Along Routes by Consortia Call Grant agreement NWA.1160.18.038 (principal investigator, M. H. Cnossen; project manager, S. H. Reitsma).
Authorship
Contribution: R.A.A. and J.J.d.V. performed the article screening; R.A.A. created the figures and wrote the paper; and all authors helped to define the research question and the search terms and provided feedback on the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
A complete list of the members of the SYMPHONY Consortium appears in “Appendix.”
Correspondence: Moniek P. M. de Maat, Department of Hematology, Erasmus MC, University Medical Center Rotterdam, PO Box 2040, 3000CA Rotterdam, the Netherlands; e-mail: m.demaat@erasmusmc.nl.
Appendix
The SYMPHONY Consortium, which aims to orchestrate personalized treatment in patients with bleeding disorders, is a unique collaboration between patients, health care professionals, and translational and fundamental researchers specialized in inherited bleeding disorders, as well as experts from multiple disciplines. It aims to identify best treatment choice for each individual based on bleeding phenotype. In order to achieve this goal, work packages (WPs) have been organized according to 3 themes: diagnostics (WPs 3 and 4), treatment (WPs 5-9), and fundamental research (WPs 10-12).
Members of the SYMPHONY Consortium: Prof.dr. C.J. Fijnvandraat, Dr. S. Gouw, Prof.dr. R.A.A. Mathôt, A. Janssen, M. Brands, L. Bukkems, M.E. Cloesmeijer, S. Koopman and Dr. L. Haverman, Amsterdam UMC, location AMC, Amsterdam, The Netherlands; Dr.ir. R. Bierings, Prof.dr. A. Lex Burdorf, Dr. M.H. Cnossen, Prof.dr. J.A. Hazelzet, Drs. E.J. Huisman, Dr. M.J.H.A. Kruip, Prof.dr. F.W.G. Leebeek, Dr. N. van Leeuwen, Dr. H.F. Lingsma, Prof.dr. M.P.M. de Maat, Dr. I. van Moort, Dr. S. Polinder, Dr. S.H. Reitsma, Mr. E. Roest, R.A. Arisz, L.G.R. Romano, W. Al Arashi, E.S. van Hoorn and M.C.H.J. Goedhart, Erasmus MC, Rotterdam, The Netherlands; Prof.dr. C.A. Uyl, Prof.dr. C.A. Uyl, Erasmus Universiteit Rotterdam, Rotterdam, The Netherlands; Prof.dr. J.G. van der Bom, Dr. M.H.A. Bos, Prof.dr. H.C.J. Eikenboom, Dr. F.J.M. van der Meer, S.N.J. Laan, LUMC, Leiden, The Netherlands; Dr. S. Schols, Radboud UMC, Nijmegen, The Netherlands; Dr. E. van den Akker, Dr. M. van den Biggelaar, Prof.dr. M. de Haas, Prof.dr. A.B. Meijer Prof.dr. J. Voorberg, J.D.C.A. Del Castillo Alferez and H. Zhang, Sanquin Research, Amsterdam, The Netherlands; Prof.dr. K. Meijer, UMC Groningen, Groningen, The Netherlands; Prof.dr. A. Bredenoord, Dr. K. Fischer, Dr. R. van der Graaf, Prof.dr. J.K. Ploos van Amstel, Dr. R.E.G. Schutgens, Dr. R.T. Urbanus, L. Baas, M. Zivkovic, UMC Utrecht, Utrecht, The Netherlands; Mr. S. de Jong, Bayer B.V.; Ms.N. Jansen, CSL Behring B.V.; Dr. M. Driessens and Dr. G. Goedhart, HemoNed; Mr. S. Meijer, Mr. W. Ninaber and Mr. J. Schipper, Netherlands Society of Haemophilia Patients (NVHP); Swedisch Orpahn Biovitrum (Sobi): Mr. J. Boender.

