

Key Point
Structural changes in coagulation FIX may be induced by recoding of the gene and could impact immunogenicity.

Abstract
Hemophilia B is a blood clotting disorder caused by deficient activity of coagulation factor IX (FIX). Multiple recombinant FIX proteins are currently approved to treat hemophilia B, and several gene therapy products are currently being developed. Codon optimization is a frequently used technique in the pharmaceutical industry to improve recombinant protein expression by recoding a coding sequence using multiple synonymous codon substitutions. The underlying assumption of this gene recoding is that synonymous substitutions do not alter protein characteristics because the primary sequence of the protein remains unchanged. However, a critical body of evidence shows that synonymous variants can affect cotranslational folding and protein function. Gene recoding could potentially alter the structure, function, and in vivo immunogenicity of recoded therapeutic proteins. Here, we evaluated multiple recoded variants of F9 designed to further explore the effects of codon usage bias on protein properties. The detailed evaluation of these constructs showed altered conformations, and assessment of translation kinetics by ribosome profiling revealed differences in local translation kinetics. Assessment of wild-type and recoded constructs using a major histocompatibility complex (MHC)-associated peptide proteomics assay showed distinct presentation of FIX-derived peptides bound to MHC class II molecules, suggesting that despite identical amino acid sequence, recoded proteins could exhibit different immunogenicity risks. Posttranslational modification analysis indicated that overexpression from gene recoding results in suboptimal posttranslational processing. Overall, our results highlight potential functional and immunogenicity concerns associated with gene-recoded F9 products. These findings have general applicability and implications for other gene-recoded recombinant proteins.
Introduction
Since the introduction of recombinant human insulin in 1982, the number of approved recombinant therapeutic proteins and their market share has grown substantially.1,2 As of 2020, there are 148 unique peptide and recombinant protein therapeutics approved by the Food and Drug Administration (FDA).3 Approximately 15% of the recombinant therapeutic proteins approved by the FDA between 2011 and 2020 are coagulation factors.2,3 An increasingly common bioengineering strategy is codon optimization. The technique takes advantage of the genetic code degeneracy to introduce synonymous codon substitutions in the therapeutic coding sequence, with the primary objective of achieving increased expression of the protein of interest.4 Multiple parameters including codon usage frequencies, messenger RNA (mRNA) structure, guanine-cytosine nucleotide content, and repeat sequences are typically taken into consideration during codon optimization.5 Recoding a gene sequence through codon optimization has been shown to improve protein expression through multiple mechanisms, including increase in the rate of transcription,6 mRNA stability,7 and/or translation rate.8 These diverse mechanisms often work in concert as these different properties are highly interconnected.9
An underlying assumption of gene recoding is that because the primary sequence of the protein remains same, the introduced variants are “silent” and do not affect protein properties. A large body of literature10-15 suggests that this assumption is incorrect. Synonymous codon changes were shown to affect protein folding/structure,13,16 ,-19 function,11 alter responses to medication,20 and cause human diseases.9,10,21 , -25 Gene recoding, which involves a large number of synonymous substitutions, was also shown to affect protein structure,26 function,27 and posttranslational modification profiles.28
A potential safety concern with therapeutic proteins is immunogenicity. Immune responses to therapeutic proteins affect their safety and efficacy and are an important concern during drug development and licensure.29 The immune responses to protein therapeutics are initiated when foreign peptides are presented to the immune system by major histocompatibility complex class II (MHC-II) molecules on the surface of antigen-presenting cells (APCs) or MHC-I molecules in the case of endogenously expressed replacement proteins in gene therapy.30 Previously, we showed that one type of recoding of coagulation factor IX (FIX) gene, F9, resulted in increased expression but led to altered conformation of FIX, potentially due to changes in local translation kinetics.26 It is plausible that conformational changes could affect proteolytic cleavage of the protein and MHC-II presentation by APCs, consequently resulting in altered immunogenicity. Overall, given that gene recoding has the potential to affect the safety of therapeutic proteins, a comprehensive evaluation of the consequences of gene recoding is needed.
In the current study, we compared multiple types of recoded F9 variants with the matched control wild type. These recoded constructs showed improved protein expression but showed altered protein conformations. We assessed the immunogenicity implications of altered conformations by employing MHC-associated peptide proteomics (MAPPs) assay in conjunction with T-cell proliferation assay. The results indicated that despite having a similar amino acid sequence, recoded F9 constructs potentially exhibit different immunogenicity risk. Additionally, assessment of posttranslational processing and specific activity of FIX indicated that overexpression of recombinant proteins could overwhelm posttranslational processing machinery and result in suboptimal processing.
Materials and methods
Plasmids
F9 constructs were synthesized and subcloned into pd608 and pd2109 plasmid vectors by ATUM (https://www.atum.bio/). Subcloning of F9 open reading frame sequences into pcDN5/FRT/V5-His Topo vector was performed by Genscript (https://www.genscript.com/). All plasmid constructs included, at the C-terminus, 48 codons encoding the V5-His tag to allow for a simplified purification scheme.
In silico analysis
The predicted secondary structure and minimum free energies of F9 constructs were obtained by using NUPACK software. The codon adaptation index values were determined as previously described.26
Cell culture
HEK293 cell line was selected for performing experiments in the study with the rationale that it is a human origin cell line that is frequently employed in recombinant therapeutic production. A liver-based cell line like the HEPG2 cell line could have better mimicked in vivo production conditions and expressed high-quality FIX,31 but our preliminary experiments showed that this cell line is difficult to transfect, and recombinant protein yields were low for purifying sufficient quantities for further characterization. Transient transfections were performed in HEK293T cells using Lipofectamine 3000 reagent (ThermoFisher). Lentiviral-transduced stable expression cell lines were generated by employing HEK293T cells, pD2109 F9 constructs, and puromycin selection. Quantitation of lentiviral vector copy numbers in selected stable expression clones was performed by using Lenti-X Provirus Quantitation Kit (Takara Bio Inc.) following the manufacturer’s instructions. For analyzing propeptide cleavage efficiency in these stable cell lines, cells were transfected with pcDNA3.1 vector encoding furin protease. Flp-In-293 stable expression cell lines were generated by cotransfecting individual pcDNA5/FRT/V5-His F9 constructs and pOG44 vector encoding recombinase and selecting cells for hygromycin resistance.
For the measurement of antigen, activity, and mRNA expression and protein purification, cells were plated in Dulbecco’s modified Eagle medium with 10% fetal bovine serum and vitamin K3 (50 ng/mL). The cell culture media was changed to OPTI-MEM (ThermoFisher) with vitamin K3 at the time of transfection or 24 hours postplating for stable expression cell lines. Supernatant media and cell lysates were harvested 24 hours following transfection or change of media to OPTI-MEM. Purification of FIX was performed by affinity tag purification using anti-V5 tag gel (MBL International) as described previously.11
Antigen and mRNA measurement
FIX antigen was assessed by immunoblotting employing mouse anti-V5 monoclonal antibody (ThermoFisher) and/or enzyme-linked immunosorbent assay (ELISA) (Affinity Biologicals). F9 mRNA expression analysis was performed by using TaqMan Gene Expression Assays (ThermoFisher) targeted against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Hs02758991_g1) and V5-His tag sequences of F9 constructs (custom made). Fold differences in mRNA expression were calculated using ΔΔCp method. All data were calculated from at least 3 biological replicate samples.
Specific activity measurement
To calculate specific activity of purified FIX samples (n = 3), FIX ELISA was calibrated against the current World Health Organization international standard for FIX antigen plasma, and 1 unit of antigen was assumed to be 5 µg/mL (ie, a normal plasma level of FIX). Activated partial thromboplastin time (aPTT) test (SynthAFax) was performed in congenital FIX-deficient plasma (HRF, Inc.) with ACL TOP 550 analyzer (Instrumentation Laboratory). Chromogenic assay (HYPHEN Biomed) was scaled down to 26 µL for 384-well microplate, otherwise following the manufacturer’s instructions. Absorbance was recorded with Biotek Synergy H4 (BioTek Instruments) at 450 nm. Maximal rate of substrate conversion (0 to 2 minutes) was used as assay readout. ELISA (Affinity Biologicals) was performed according to the manufacturer’s instructions. In-house thrombin generation test (TGT) was performed as follows: congenital FIX-deficient citrated plasma (50% vol/vol) was mixed with corn trypsin inhibitor (100 μg/mL, Haematologic Technologies), fluorogenic substrate Z-Gly-Gly-Arg-7-Amino-4-methylcoumarin (AMC) (800 μM, Bachem), phospholipid vesicles (4 μM, Rossix), and tissue factor (0.35 pM, Innovin, Baxter). Plasma was activated by 10 mM CaCl2 and 10 mIU XIa (Reference Reagent, code 11/236) and immediately mixed with samples (25% vol/vol). 7-Amino-4-methylcoumarin fluorescence was recorded at 37°C and 460 nm. Thrombin peak height was used as assay readout.
Potency calculation was performed in OriginLab software (ver. 2020b), as described previously,32 as an average for 3 to 6 dilutions of each FIX sample against a calibration curve of 7 points. The specific activity and its standard deviation were estimated using bivariate first-order Taylor expansion.33,34
CD spectropolarimetry
Circular dichroism (CD) analysis of FIX was performed using J-810 CD Spectrophotometer (Jasco). Circular dichroism readings were measured from pooled purified protein from 3 preps each of wild-type and recoded FIX samples placed in 1 mm path-length absorption cuvettes (Hellma Analytics) at a constant temperature of 4°C and far-UV wavelength range of 190 to 260 nm. Protein samples were run at a concentration of 370 µg/mL in phosphate (4 mM)-buffered saline (155 mM) solution. The low phosphate concentration introduced minimal noise in the CD spectra. Significant noise was noted at 197 nm and below.
FIX inhibitory antibody assay
FIX-depleted plasma supplemented with polyclonal anti-FIX antibodies (Affinity Biologicals), hemophilia B patient plasma with anti-FIX inhibitory antibodies, or normal human plasma (National Institutes of Health Blood Bank) were heat inactivated at 56°C for 30 minutes and centrifuged to remove precipitates. FIX samples (n = 3) at 20 ng/mL concentration were incubated with serial dilutions of the plasma at 30°C for 1 hour.26 FIX activity was then measured in 3 independent experiments by chromogenic assay (Biophen Factor IX, Aniara). Half-max (ED50) values were calculated using 4-parameter dosage response curves stratified by treatment type.
Posttranslational modifications analysis
Limited proteolysis
Limited proteolysis with trypsin was performed as described previously.14 Briefly, pooled purified protein from 3 preps each of wild-type and recoded FIX samples were digested with a range of concentrations of trypsin (0-0.000125 mg/mL) for 3 minutes at 37°C. Digestion reaction was terminated by the addition of 4X LDS buffer with dithiothreitol and boiling samples for 20 minutes at 100°C. Samples were analyzed by immunoblotting using mouse anti-V5 monoclonal antibody.
Ribosome profiling
Ribosome profiling experiments and data analysis were performed as described previously,26,36 with few modifications. Briefly, library construction was conducted using the Illumina TruSeq Ribo Profile (Mammalian) Kit per the manufacturer’s instructions, with modifications in harvest (cells were flash frozen immediately after removal of media), RNA isolation/purification (isopropanol isolation was used to improve the yield), and ribosome-protected fragment (RPF) size selection (∼20-32 nucleotide). Ribosome profiling and RNA sequencing libraries were sequenced using Illumina HiSeq2500.
RPF fragments of 20 to 22 and 27 to 29 nucleotides in length were used for analysis with an A-site offset of 15 nucleotides. Normalized codon coverage for the coding sequences of F9 and control genes GAPDH and ACTB was calculated as (#RPFs with codon X in A site/average #RPFs from coding sequence). Data were then averaged across the replicates (n = 5) to generate final RPF coverage plots. Normalized RPF coverage for the coding sequences of each gene was plotted using A-site fragment density per codon.
MAPPs assay
The study was performed by PROIMMUNE (https://www.proimmune.com/) as previously described.37 In brief, monocytes were isolated from peripheral blood samples of 6 donors by positive selection and differentiated in vitro into immature monocyte-derived dendritic cells (MoDCs). The immature MoDCs were cultured and matured in the presence of FIX at 50 µg/mL concentration. The mature MoDCs were harvested and lysed, and HLA class II DR molecules were recovered using immune-affinity columns. The HLA class II DR–bound peptides were then analyzed by high-resolution-sequencing mass spectrometry, and the resulting data were analyzed using sequence analysis software referencing the Swiss-Prot Human Proteome Database. HLA-DRB1 typing information of the donors is provided in supplemental Table 1. Pooled purified FIX samples from ≥11 preps were used in the assay.
T-cell proliferation assay
The assay was performed by PROIMMUNE. In brief, peptides were synthesized and incubated with peripheral blood mononuclear cells (40 donors) at 5 µM concentration in 6 replicates. Four control antigens were included in the assays: tuberculin-purified protein derivative, keyhole limpet hemocyanin, hemagglutinin, and tetanus toxoid. Untreated control wells were included in each plate. Cultures were incubated for 7 days before measuring proliferation, using a carboxyfluorescein diacetate succinimidyl ester staining assay. The percentage of stimulation above background was measured for each peptide/donor combination. HLA-DRB1 typing information of the donors was provided in supplemental Table 1.
Statistical analysis
The Kolmogorov-Smirnov test was used to compare curves of cumulative sum data of ribosome profiling experiments. The Kruskal-Wallis test was used to compare antigen expression data among constructs. Pairwise comparison between groups or individual constructs was performed with the Wilcoxon signed-rank test, using Benjamini-Hochberg multiple hypothesis correction. Specific activity differences between FIX constructs were measured by using a 1-sample t test on log2-transformed fold changes to test for a mean of 0. The R programming language, Dose Response Curves package was used to fit FIX activity inhibition against dilution. Then, models were compared using the χ2 test on the difference in their loglikelihoods.
Results
Gene recoding resulted in increased expression and lower specific activity
We generated 6 recoded F9 constructs to systematically explore Codon Adaptation Index (CAI),38 a coding sequence variable here defined as “high” (OPT-1; 1A, 1B, and 1C) and “low” (OPT2; 2A, 2B, and 2C) (Figure 1A). Additionally, we also explored similarity to wild-type sequence, here defined as “close” (86% for 1B, 90% for 2B), “medium” (79% for 1A, 80% for 2A), and “distant” (73% for both 1C and 2C) (Figure 1A; supplemental Table 2). The 15 N-terminal codons were not recoded to reduce the risk that events following initiation of translation would impact the effect of recoding on the rest of the sequence.39
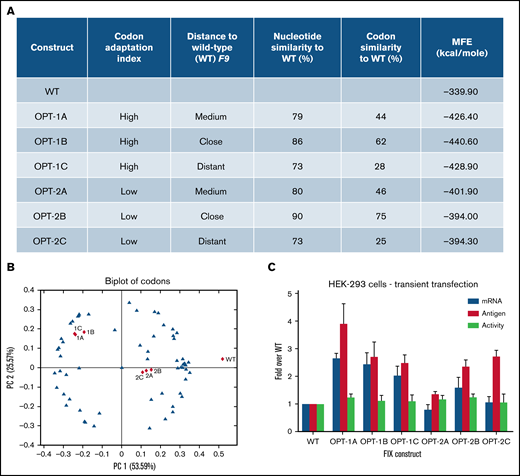
Codon usage variable and properties of wild-type and recoded F9 sequences. (A) The underlying primary codon usage variables, namely CAI and similarity to wild-type sequence employed in the design of recoded F9 constructs. Constructs were designed to have either “high” (1A, 1B, and 1C) or “low” (2A, 2B, and 2C) CAI and “close” (1B and 2B), “medium” (1A and 2A), or “distant” (1C and 2C) similarity to wild-type sequence. Percentages of nucleotides and codons altered in recoded sequences and in silico–predicted minimum free energies (MFE) of resulting mRNA structures were also shown. (B) A biplot of codons of wild-type and recoded F9 sequences showing the segregation of sequences based on codon adaptation indices of constructs. (C) Antigen and mRNA expression data and activity levels of wild-type and recoded F9 sequences in transient transfection experiments. Recoded constructs demonstrated higher levels of F9 mRNA and FIX antigen expression. Data are represented as mean ± standard deviation. WT, wild-type.
Codon usage variable and properties of wild-type and recoded F9 sequences. (A) The underlying primary codon usage variables, namely CAI and similarity to wild-type sequence employed in the design of recoded F9 constructs. Constructs were designed to have either “high” (1A, 1B, and 1C) or “low” (2A, 2B, and 2C) CAI and “close” (1B and 2B), “medium” (1A and 2A), or “distant” (1C and 2C) similarity to wild-type sequence. Percentages of nucleotides and codons altered in recoded sequences and in silico–predicted minimum free energies (MFE) of resulting mRNA structures were also shown. (B) A biplot of codons of wild-type and recoded F9 sequences showing the segregation of sequences based on codon adaptation indices of constructs. (C) Antigen and mRNA expression data and activity levels of wild-type and recoded F9 sequences in transient transfection experiments. Recoded constructs demonstrated higher levels of F9 mRNA and FIX antigen expression. Data are represented as mean ± standard deviation. WT, wild-type.
Principal component analysis of all 64 codons (blue triangles) and recoded sequences (red triangles) (Figure 1B) revealed grouping of OPT-1 and OPT-2 constructs together, confirming the inherent similarity in the CAI design principles among each group of constructs. In silico analysis of the predicted mRNA structures revealed altered secondary structure (supplemental Figure 1) and lower minimum free energy, an indicator of the increased structural stability of the recoded constructs (Figure 1A). In transient transfections, all recoded constructs showed increased FIX expression (Figure 1C). Antigen expression analysis of constructs stratified by high (OPT-1) or low (OPT-2) CAI values identified significant differences (P = .0015) (supplemental Figure 2A). A pairwise comparison revealed significant differences between all groups (P < .045). Antigen expression of constructs stratified by distance to wild-type sequence (close, “A”; medium, “B”; and distant, “C”) also identified significant differences (P = .0157) (supplemental Figure 2B). Pairwise comparison showed significant differences between wild-type and all other groups (P = .015 for all comparisons); however, differences between A, B, and C groups were not significant. This data indicated CAI as the primary variable underlying significant differences in antigen expression from recoded F9 constructs. Contrary to the differences in antigen expression, activity assessment by chromogenic assay revealed relatively similar values for all constructs (Figure 1C). This data indicated lower specific activity of recoded constructs, which is probably a combined result of overexpression from gene recoding and complex posttranslational modifications that FIX has to undergo. The mRNA levels of individual F9 constructs were in agreement with their respective antigen levels (Figure 1C), suggesting a primary role of transcription and/or mRNA stability in the increased FIX expression of the recoded constructs.
Gene recoding resulted in altered conformation
For the experimental feasibility, we evaluated the effects of recoding using 2 recoded constructs, OPT-1A and OPT-2A, both carrying an intermediate level of CAI and nucleotide/codon similarity to the wild-type and representing the highest(OPT-1A) and lowest (OPT-2A) levels of FIX expression among recoded constructs (Figure 1C). Quantitation of lentiviral copy numbers in the clones selected for comparable expression revealed expression of FIX from 4.17 ± 0.24 copies for wild-type, 3.02 ± 0.17 copies for OPT-1A, and 1.49 ± 0.2 copies for OPT-2A. The apparent discrepancy in the observed and expected antigen levels based on integrated copy numbers and expression levels in transient transfection experiments could be due to chromosomal position effects on expression from randomly integrated transgene copies in these cell lines.40,41 The F9 mRNA expression levels within the selected individual clones were comparable (Figure 2A). Recombinant FIX purified from these selected clones was used for the analysis of protein conformation, specific activity, and posttranslational modifications.

Characterization of recoded FIX constructs expressed from lentiviral transduction stable expression system. (A) The mRNA and antigen expression levels of wild-type (WT), OPT-1A, and OPT-2A variant lentiviral-transduced stable expression clones selected for further characterization. Data are represented as mean ± standard deviation (SD). (B) CD analysis of FIX secondary structures. Recoded FIX constructs showed similar CD spectra profiles that are distinct from WT FIX. (C) Limited trypsin digestion profiles of WT, OPT-1A, and OPT-2A constructs. Both OPT-1A and OPT-2A constructs showed increased sensitivity to trypsin digestion compared with WT FIX (highlighted by red color squares). (D-E) Inhibitory antibody assay data from assays employing FIX-depleted plasma spiked with polyclonal anti-FIX antibodies and hemophilia B patient with anti-FIX inhibitory antibodies, respectively. Data showed significantly different inhibition kinetics for OPT-1A and OPT-2A FIX in comparison with wild type when incubated with the spiked plasma (panel D, with ED50 0.0284, 0.0339, and 0.0286 for WT, OPT-1A, and OPT-2A) and hemophilia B patient plasma (panel E, with ED50 0.0637, 0.0580, and 0.0453 for WT, OPT-1A, and OPT-2A), respectively. Together, data in panels B-E demonstrated conformational differences of recoded FIX constructs relative to WT FIX. (F) Specific activity assessment data for recoded FIX constructs presented as fold activity over WT. Data are represented as mean ± SD. (G) Propeptide processing and γ-carboxylation profiles of purified FIX from selected clones.
Characterization of recoded FIX constructs expressed from lentiviral transduction stable expression system. (A) The mRNA and antigen expression levels of wild-type (WT), OPT-1A, and OPT-2A variant lentiviral-transduced stable expression clones selected for further characterization. Data are represented as mean ± standard deviation (SD). (B) CD analysis of FIX secondary structures. Recoded FIX constructs showed similar CD spectra profiles that are distinct from WT FIX. (C) Limited trypsin digestion profiles of WT, OPT-1A, and OPT-2A constructs. Both OPT-1A and OPT-2A constructs showed increased sensitivity to trypsin digestion compared with WT FIX (highlighted by red color squares). (D-E) Inhibitory antibody assay data from assays employing FIX-depleted plasma spiked with polyclonal anti-FIX antibodies and hemophilia B patient with anti-FIX inhibitory antibodies, respectively. Data showed significantly different inhibition kinetics for OPT-1A and OPT-2A FIX in comparison with wild type when incubated with the spiked plasma (panel D, with ED50 0.0284, 0.0339, and 0.0286 for WT, OPT-1A, and OPT-2A) and hemophilia B patient plasma (panel E, with ED50 0.0637, 0.0580, and 0.0453 for WT, OPT-1A, and OPT-2A), respectively. Together, data in panels B-E demonstrated conformational differences of recoded FIX constructs relative to WT FIX. (F) Specific activity assessment data for recoded FIX constructs presented as fold activity over WT. Data are represented as mean ± SD. (G) Propeptide processing and γ-carboxylation profiles of purified FIX from selected clones.
To assess protein conformational differences, we employed CD spectropolarimetry, limited proteolysis, and FIX inhibitory antibody assays. The secondary structure analysis of 2 recoded constructs (OPT-1A and OPT-2A) using CD spectropolarimetry showed nearly overlapping CD profiles (similar within ∼1 mdeg) that were distinct from that of wild-type (Figure 2B). Limited proteolysis by trypsin revealed increased sensitivity of recoded constructs compared with wild-type (Figure 2C). Inhibitory antibody assays were performed in 2 formats. In the first, FIX-depleted plasma was spiked with polyclonal anti-FIX antibodies, and inhibition of FIX activity was monitored. In the second, hemophilia B plasma with FIX inhibitory antibodies was used. The OPT-1A exhibited significantly different (P = .0029) inhibition kinetics, represented as ED50 values, from the wild-type when incubated with the spiked plasma (Figure 2D), whereas the OPT-2A exhibited significantly different (P = .00062) inhibition kinetics from the wild-type when incubated with patient plasma having inhibitory anti-FIX antibodies (Figure 2E). In addition, OPT-1A and OPT-2A showed significantly different (P = .012 and .0042, respectively) inhibition kinetics as compared with each other. Together, these results indicated conformational differences between the constructs under study.
We compared the specific activity of FIX variants using 3 activity assays: chromogenic assay, aPTT and TGT test, and an ELISA-based antigen assay (Figure 2F). In this analysis, OPT-2A showed relatively higher specific activity (≥22% higher than wild-type), followed by OPT-1A (≥12% higher than wild-type), in all 3 assays. The observed differences are statistically significant for wild-type vs OPT-1A measured by chromogenic and TGT (P = .01) assays and wild-type vs OPT-2A measured by chromogenic (P = .018) assay. These specific activity differences seem to be partially explained by relatively lower propeptide levels and higher γ-carboxyglutamic acid (Gla) content observed in OPT-1A and OPT-2A clones, respectively (Figure 2G). We also observed assay-based differences in the measurement of specific activities, where TGT consistently showed lower absolute specific activities (supplemental Figure 3). These absolute specific activities of our samples varied between 252 and 461 IU/mg (based on aPTT clotting assay). This is comparable to the specific activity of the recombinant FIX product BeneFIX, which has the typical specific activity of “greater than or equal to 200 IU per milligram of protein” (see DailyMed - BENEFIX [Coagulation Factor IX Recombinant Ki] [nih.gov]).
In the posttranslational modifications analysis, wild-type, OPT-1A, and OPT-2A constructs showed propeptide levels of 4.8%, 2.8%, and 4.3% and Gla content of 10.5, 10.4, and 11 Gla per mol, respectively (Figure 2G; supplemental Table 3). For comparison, plasma-derived FIX exhibits nondetectable propeptide sequence and 12 Gla per mol.42 The presence of propeptide sequences and relatively lower Gla of FIX derived from stable expression cell lines in our study suggested that the endogenous γ-glutamyl carboxylase and furin protease activity needed for γ carboxylation and propeptide cleavage was not sufficient to optimally process the overexpressed FIX. In support of this postulate, when furin was coexpressed in these stable expression cell lines, FIX with propeptide sequences was not detected (supplemental Figure 4). We did not examine the effects of vitamin K epoxide reductase coexpression in the present work, though literature predicts this may need to be titrated carefully in conjunction with the γ-glutamyl carboxylase.43 Other evaluated posttranslational modifications including S53 O-glycosylation, D64 and K91 hydroxylations, N199 deamidation, and M391 oxidation were largely similar between the constructs (supplemental Table 3).
Ribosome profiling revealed differences in translational kinetics
We used ribosome profiling to assess potential translation kinetics differences due to recoding.44 Ribosome profiling allows monitoring of translation at codon-level resolution through sequencing of the actively translating transcriptome. This technique involves isolation and deep sequencing of mRNA fragments that are protected from ribonuclease digestion by translating ribosomes. Ribosomes are associated with codons that are translated slowly for longer periods, and these RPFs are overrepresented in the ribosome profiling data. We assessed the translation of F9, as well as ACTB and GAPDH, as controls. The RPF profiles of wild-type and recoded F9 were presented as cumulative sums of normalized and log-transformed RPF data. In this analysis, the RPF profiles for both OPT-1A and OPT-2A were significantly different from the RPF profile of wild-type (P = 4.87e-04 and P = 2.211e-08 for OPT-1A and OPT-2A, respectively) (Figure 3A). Additionally, all 3 constructs showed distinct profiles in the region of sequences encoding activation peptide and peptidase domains. Importantly, the RPF profiles in the identical C-terminal V5-His tag regions of F9 constructs (codons 463-509) (Figure 3A) and control genes ACTB (Figure 3B) and GAPDH (Figure 3C) were comparable. Together, this data demonstrated the differences in local translational kinetics of the wild-type and recoded F9 constructs.
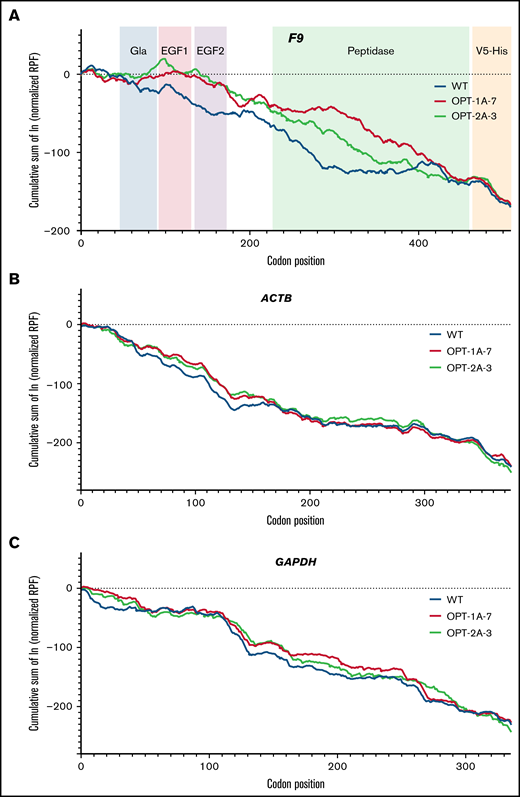
RPF distribution along F9, ACTB, and GAPDH genes. The cumulative sum of normalized and log-transformed RPF density along the F9 (A), ACTB (B), and GAPDH (C) genes in lentiviral-transduced stable expression cell lines of wild-type (WT), OPT-1A, and OPT-2A constructs is shown. FIX domain structure is included in panel A to illustrate the local translation kinetics differences. (A) Distinct RPF profiles for both OPT-1A and OPT-2A variants compared with WT. On the other hand, the RPF profiles in the identical C-terminal V5-His tag regions of F9 constructs (A) and control genes ACTB (B) and GAPDH (C) are similar.
RPF distribution along F9, ACTB, and GAPDH genes. The cumulative sum of normalized and log-transformed RPF density along the F9 (A), ACTB (B), and GAPDH (C) genes in lentiviral-transduced stable expression cell lines of wild-type (WT), OPT-1A, and OPT-2A constructs is shown. FIX domain structure is included in panel A to illustrate the local translation kinetics differences. (A) Distinct RPF profiles for both OPT-1A and OPT-2A variants compared with WT. On the other hand, the RPF profiles in the identical C-terminal V5-His tag regions of F9 constructs (A) and control genes ACTB (B) and GAPDH (C) are similar.
Gene recoding alters peptide presentation by MHC-II on APCs
Given that recoded FIX revealed altered conformations, we investigated whether these variants may be processed and presented by the immune system differently than the wild type. A MAPPs assay45 was performed using MoDCs from 6 donors. Overall, 7 FIX-specific peptides were identified on MoDCs (Figure 4A). As expected, MHC-II molecules from each donor presented a distinct set of peptides, and no donor presented all of the identified peptides. Interestingly, peptide 4 (amino acids 307-325) was presented from MoDCs of donor D1890 when exposed to OPT-1A and of D1891 when exposed to OPT-1A and OPT-2A, but no donors presented these peptides when incubated with wild type. Similarly, there were peptides that were presented by MoDCs incubated with wild type but not with some of the recoded proteins. Further, we explored the potential capacity of the identified peptides to induce T-cell proliferation. In this analysis, several of the peptides tested were able to induce CD4+ cell proliferation (Figure 4B; supplemental Table 5), with peptide 6 inducing proliferation in more peripheral blood mononuclear cell samples than other peptides.
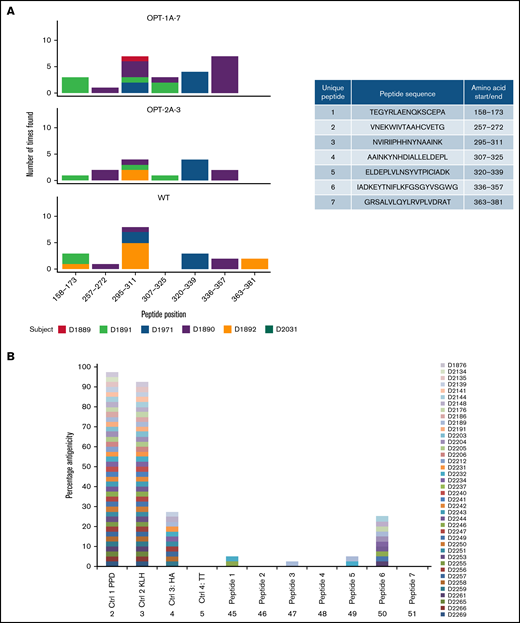
Immunogenicity of recoded FIX constructs. Immunogenicity assessment of recoded FIX constructs by MAPPs assay and T-cell proliferation assay is shown. (A) Distinct presentation of FIX-derived peptides bound to MHC-II molecules by wild-type (WT) and recoded FIX constructs. The sequences of the FIX peptides presented by MoDCs and their amino acid positions are included in the side table. The figure shows the number of times each of the peptides were presented by MoDCs, colored differently for each donor source. (B) Potential immunogenicity of these peptides as suggested by CD4+ T-cell proliferation in assay. Data are shown as percentage of donors showing response to test antigens measured as stimulation above background (≥0.5% and 2 standard errors greater than background). Four control antigens, tuberculin-purified protein derivative (PPD), keyhole limpet hemocyanin (KLH), hemagglutinin (HA), and tetanus toxoid (TT), were included in the assay.
Immunogenicity of recoded FIX constructs. Immunogenicity assessment of recoded FIX constructs by MAPPs assay and T-cell proliferation assay is shown. (A) Distinct presentation of FIX-derived peptides bound to MHC-II molecules by wild-type (WT) and recoded FIX constructs. The sequences of the FIX peptides presented by MoDCs and their amino acid positions are included in the side table. The figure shows the number of times each of the peptides were presented by MoDCs, colored differently for each donor source. (B) Potential immunogenicity of these peptides as suggested by CD4+ T-cell proliferation in assay. Data are shown as percentage of donors showing response to test antigens measured as stimulation above background (≥0.5% and 2 standard errors greater than background). Four control antigens, tuberculin-purified protein derivative (PPD), keyhole limpet hemocyanin (KLH), hemagglutinin (HA), and tetanus toxoid (TT), were included in the assay.
Limited expression improves FIX processing
To better understand the undue pressure exerted by FIX overexpression on the posttranslational processing machinery, we established stable expression cell lines of wild-type, OPT-1A, and OPT-2A constructs using the Flp-In-293 cell line that expresses the protein of interest from a single copy, integrated at a prespecified genomic location.
In the Flp-In-293 system, the wild type and OPT-2A showed comparable FIX expression, whereas OPT-1A showed about 10-fold higher expression (Figure 5A). The relative mRNA levels in cell lines corresponded with the differences in FIX expression levels (Figure 5A). The differences in expression levels identified in the Flp-In-293 system could be true differences that are attributable to the recoding because variables like copy number per cell and site of integration are controlled. In posttranslational modifications analysis, wild-type, OPT-1A, and OPT-2A constructs showed propeptide levels of 0.5%, 3.2%, and 0.7% and total Gla content of 11.85, 11.23, and 11.83 Gla per mol, respectively (Figure 5B; supplemental Table 4). These numbers, specifically Gla content, indicated a relatively enhanced posttranslational processing of FIX expressed from the Flp-in-293 cell lines compared with those expressed from lentiviral-transduced stable expression cell lines (Figure 2G). Moreover, posttranslational processing of FIX constructs seemed to be affected by respective expression levels, where OPT-1A with the highest expression showed relatively lower posttranslational processing. Subsequently, OPT-1A showed relatively lower specific activity in all 3 activity assays (Figure 5C). However, these differences are statistically significant only for wild-type vs OPT-1A comparison measured by chromogenic assay (P = .039). Other analyzed posttranslational modifications were largely similar, with no discernible differences (supplemental Table 4).
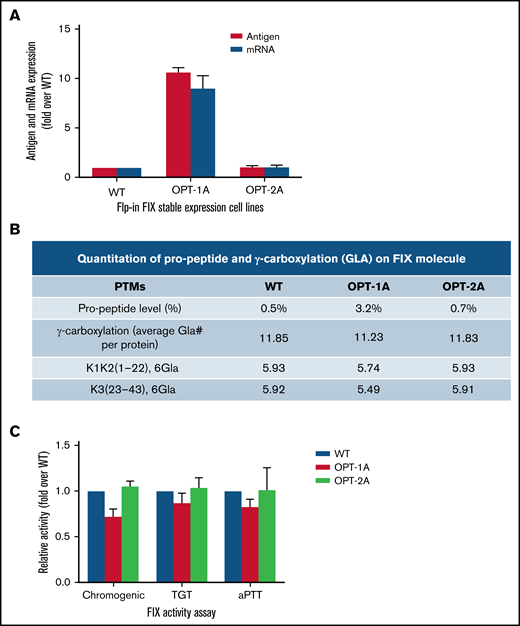
Characterization of recoded FIX constructs expressed from Flp-In system. (A) The mRNA and antigen expression levels of wild-type (WT), OPT-1A, and OPT-2A variants from single copy expression Flp-in system stable expression cell lines. Data are represented as mean ± standard deviation (SD). (B) Propeptide processing and γ-carboxylation profiles of purified FIX. (C) Specific activity data for recoded FIX constructs presented as fold activity over WT. Data are represented as mean ± SD. Data in panels B and C demonstrated relatively higher propeptide FIX fraction, lower Gla, and lower specific activity for FIX expressed from the variant with the highest antigen expression, OPT-1A. This data suggested suboptimal posttranslational processing of FIX when overexpressed.
Characterization of recoded FIX constructs expressed from Flp-In system. (A) The mRNA and antigen expression levels of wild-type (WT), OPT-1A, and OPT-2A variants from single copy expression Flp-in system stable expression cell lines. Data are represented as mean ± standard deviation (SD). (B) Propeptide processing and γ-carboxylation profiles of purified FIX. (C) Specific activity data for recoded FIX constructs presented as fold activity over WT. Data are represented as mean ± SD. Data in panels B and C demonstrated relatively higher propeptide FIX fraction, lower Gla, and lower specific activity for FIX expressed from the variant with the highest antigen expression, OPT-1A. This data suggested suboptimal posttranslational processing of FIX when overexpressed.
Discussion
When genetic sequences are recoded with the primary objective of enhancing the protein expression of protein therapeutics, manufacturing controls and/or downstream purification steps can most often weed out egregious effects of recoding by selecting out incorrectly folded or processed protein forms. This purification is not possible in the case of constructs used in gene therapy, for which there is no such quality control. Potential safety issues such as increased immunogenicity or toxicity driven by protein aggregation are not clear, and this risk becomes more germane as gene therapy products are increasingly a part of drug development pipelines. In this study, we focused on how gene recoding can affect properties of a therapeutic protein, including immunogenicity.
Recombinant FIX, which is used clinically to treat hemophilia B, was chosen as a model protein, mostly because the high volume of therapeutics including gene therapy applications. From a panel of F9-recoded constructs, we synthesized, purified, and extensively characterized wild-type and selected recoded variants (OPT-1A and OPT-2A). As recoding often results in greatly augmented levels of protein synthesis, it is postulated that cellular machinery involved in posttranslational processing and quality control can be overwhelmed. We therefore established multiple clones and selected those with equivalent FIX expression. In cells expressing equivalent levels of FIX, we found largely comparable posttranslational modifications. However, all 3 recombinant clones showed a lower Gla content compared with plasma-derived FIX, a critical posttranslational modification for FIX activity. These clones moreover produced FIX with conformational differences between the wild-type and recoded constructs as well as between the 2 recoded variants. These conformational differences were confirmed by 3 orthogonal techniques. These results suggested that even when controlling for the level of expression, synonymous codon changes may alter protein conformation. Further, using ribosome profiling, we could demonstrate significant translation kinetics differences among wild-type and recoded variants of FIX. These results are consistent with other studies16,17,26,46 showing that altering codon usage changes translation kinetics.
The consequences of altered conformation, potential aggregation, etc of recoded constructs are potential pitfalls but to date have not been associated with clinical risk. However, if the altered conformation due to recoding increases the immunogenicity risk of the protein therapeutic, it reflects a potential safety issue. As both wild-type and recoded proteins have the same primary sequence, it appears unlikely that these variants would elicit different immune responses. However, in the last decade, accumulating evidence37,47 -51 suggests otherwise. Recently, a prospective randomized clinical control trial demonstrated that recombinant FVIII products, despite sharing the same amino acid sequence, are more immunogenic than plasma-derived FVIII products.51 Glycosylation differences, association with plasma von Willebrand factor, and presence of cooccurring immunomodulatory proteins were cited as potential reasons behind these observed differences.51 Further analysis by MAPPs assay revealed that plasma-derived FVIII presents fewer peptides than recombinant FVIII in the presence of plasma-derived von Willebrand factor, suggesting that the 2 products are processed differently.37,52-54 We carried out a similar analysis using the wild-type and 2 recoded variants. In this study, it is clear that the wild-type and recombinant variants present different sets of FIX-derived peptides on APCs from the same donor. With this small dataset, it is impossible to determine whether recoded proteins present a greater (or lesser) risk of immune response. Our data nonetheless establishes differential processing of wild-type and recoded variants by the immune machinery of APCs.
Overexpression from multiple copies and/or recoding in lentiviral transduction expression system resulted in suboptimal posttranslation processing, specifically propeptide cleavage and γ-carboxylation that are critical for FIX activity. It is to be noted here that an extraneous source of vitamin K, which plays an important role in the γ-carboxylation, was provided to cells in the current study. Additionally, coexpression of furin improved propeptide processing. These findings agree with previous studies that indicated the requirement of furin coexpression for optimal propeptide cleavage of overexpressed FIX.43,55,56 On the other hand, controlled expression of FIX from a single copy resulted in improved posttranslational processing of the wild-type as well as the recoded constructs. Primary hepatocytes, the targeted cells of FIX expression in gene therapy are expected to better process in vivo–expressed FIX.31 However, gene recoding will result in nonphysiological expression of FIX on a per cell basis and could still overcome the intrinsic quality control mechanisms. Further studies are needed to better understand the impact of nonphysiological expression of FIX in in vivo physiological conditions. Overall, our results indicate a limited capacity of posttranslational machinery to optimally process overexpressed proteins, specifically for proteins that undergoes complex posttranslational modifications.
Another interesting observation is assay-based differences in the specific activity measurement, where TGT yielded lower measurements compared with chromogenic and aPTT assays. A recent study by Rosen et al.57 identified higher specific activity of recombinant FVIII in human plasma following gene therapy when the activity was measured by the aPTT-based assay vs the chromogenic assay. The mechanisms behind factor activity assay discrepancies remain unknown. Data on FIX activity assay consistency is not yet available for gene therapies based on wild-type FIX, although this topic is gaining more attention with the introduction of FIX Padua–based therapies. Among the FVIII gene therapy trials, similar type of discrepancies appeared across several trials that have all used codon optimization of their cassettes. This may indicate that codon optimization per se may not be an important factor or the only factor to account for these discrepancies. More data are needed to ascertain the codon optimization–specific effects related to these discrepancies. Differences in posttranslational modifications, probable protein-folding differences from codon optimization, differences in affinity to von Willebrand Factor, ectopic expression of FVIII from hepatocytes, and molecular differences between nonnative analytes and native calibrators and reagent disparities were among the suggested potential contributing factors.57,58 Overall, our results reinforce the FDA’s recommendation to validate factor activity assays for their suitability to analyze the protein of interest.59
Taken together, our study highlighted the gene recoding–induced changes in the conformation, posttranslational modifications, and function of therapeutic proteins using coagulation factor IX as a model. These changes have the potential to affect the efficacy and safety of the recombinant proteins. Such concerns may be particularly relevant to gene therapies that are typically based on recoded expression constructs and rely on intrinsic quality control mechanisms.
Acknowledgments
This work was supported by CBER operating funds and in part supported by National Heart, Lung, and Blood Institute (NHLBI) grant HL151392 (A.A.K.), NHLBI grant R01HL155986 (T.K.), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grant R01DK058702 (T.K.), and National Institutes of Health (NIH) grant R01HL128119 (T.K.).
Authorship
Contribution: C.K.-S., A.A.K., and R.C.H. conceptualized the study; C.K.-S., A.A.K., Z.E.S., R.C.H., U.K.K., A.A., G.K.H., T.K., R.P., M.V.O., and D.I.F. contributed to methodology and resources; U.K.K., A.A., G.K.H., N.H.-K., J.M.K., B.L., L.A.P., and Q.L. contributed to investigation, data curation, and formal analysis; J.M.K., J.R.M., D.D.H., J.C.A., and H.B. contributed to software and statistical analysis; U.K.K. and Z.E.S. wrote the original draft; and all authors analyzed the data, revised the manuscript, were engaged in commenting on the manuscript, and read and approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Chava Kimchi-Sarfaty, Division of Plasma Protein Therapeutics, Office of Tissues and Advanced Therapies, Center for Biologics Evaluation & Research, US Food and Drug Administration, Silver Spring, MD 20993; e-mail: chava.kimchi-sarfaty@fda.hhs.gov.

