

Key Points
The combination of FACS for enrichment of CD34+ PB cells and deep sequencing enables detection of MRD at levels down to 1:106.
NGS in CD34+ PB cells improved early prediction of molecular relapse compared with NGS of unsorted PB or CD34+ donor chimerism.

Abstract
Monitoring of measurable residual disease (MRD) in patients with acute myeloid leukemia (AML) is predictive of disease recurrence and may identify patients who benefit from treatment intensification. Current MRD techniques rely on multicolor flow cytometry or molecular methods, but are limited in applicability or sensitivity. We evaluated the feasibility of a novel approach for MRD detection in peripheral blood (PB), which combines immunomagnetic preenrichment and fluorescence-activated cell sorting (FACS) for isolation of CD34+ cells with error-reduced targeted next-generation sequencing (NGS). For clinical validation, we retrospectively analyzed 429 PB and 55 bone marrow (BM) samples of 40 patients with AML or high-risk MDS, with/without molecular relapse based on CD34+ donor chimerism (DC), in complete remission after allogeneic stem cell transplantation. Enrichment of CD34+ cells for NGS increased the detection of mutant alleles in PB ∼1000-fold (median variant allele frequency, 1.27% vs 0.0046% in unsorted PB; P < .0001). Although a strong correlation was observed for the parallel analysis of CD34+ PB cells with NGS and DC (r = 0.8601), the combination of FACS and NGS improved sensitivity for MRD detection in dilution experiments ∼10-fold to levels of 10−6. In both assays, MRD detection was superior using PB vs BM for CD34+ enrichment. Importantly, NGS on CD34+ PB cells enabled prediction of molecular relapse with high sensitivity (100%) and specificity (91%), and significantly earlier (median, 48 days; range, 0-281; P = .0011) than by CD34+ DC or NGS of unsorted PB, providing additional time for therapeutic intervention. Moreover, panel sequencing in CD34+ cells allowed for the early assessment of clonal trajectories in hematological complete remission.
Introduction
For patients with hematological malignancies such as acute myeloid leukemia (AML) or high-risk myelodysplastic syndrome (MDS), allogeneic hematopoietic stem cell transplantation (allo-HSCT) often remains the only curative treatment option.1 Nevertheless, relapse after allo-HSCT occurs in 30% to 70% of patients with AML and is the major cause of treatment failure, with dismal prognosis and a 2-year survival of <20%.2 To predict imminent relapse and to identify patients who could benefit from early therapeutic intervention, current surveillance strategies recommended by the European LeukemiaNet (ELN) include the assessment of measurable residual disease (MRD) during long-term follow-up of patients in hematological complete remission (CR).3,4
The cytomorphology-based threshold of CR is defined by <5% myeloblasts in the bone marrow (BM), and MRD refers to the presence of residual leukemic cells at frequencies of 1:103 to 1:106 of white blood cells (WBCs).4 Several studies demonstrated the prognostic value of MRD detection in AML, using established methods such as multiparameter flow cytometry (MFC)5,6 and quantitative polymerase chain reaction (qPCR).7,8 Because of the high sensitivity (up to 10−6), qPCR-based approaches are currently considered the gold standard for MRD monitoring, but are limited to ∼40% of patients with AML with specific genetic aberrations, such as NPM1 mutations or various fusion genes.4,7,8 In contrast, emerging technologies such as next-generation sequencing (NGS) can detect all types of leukemia-specific molecular alterations and hence potentially enables assessment of MRD in most patients with AML.4,9 However, in current clinical practice, most assays for genetic MRD monitoring still necessitate the analysis of BM aspirates to achieve sufficient sensitivity.
In patients undergoing allo-HSCT, the analysis of CD34+ donor chimerism (DC) in peripheral blood (PB) represents an alternative and clinically proven method for sensitive MRD detection after transplantation, which is applicable to >80% of patients with AML (with blast cells expressing the CD34 antigen) and can predict impending relapse with a latency of ∼8 weeks.10-12 More recently, MRD-guided preemptive treatment with azacitidine (AZA), monitored by CD34+ DC in PB, was reported to substantially delay or prevent hematological relapse in MDS and patients with AML.13,14 When compared with standard chimerism analysis of whole-blood samples, chimerism analysis in sorted CD34+ cells takes advantage of fluorescence-activated cell sorting (FACS) to enable the initial enrichment of the hematological stem cell compartment (which also contains potential leukemic stem cells) and subsequent short tandem repeat (STR) analysis for the detection of residual recipient cell populations.12,13 Using this technique, researchers have demonstrated the detection of 1 leukemic cell in at least 40 000 WBCs.12 However, assuming a frequency of ∼0.1% CD34+ cells in PB under normal conditions and a sensitivity of 1% to 5% for STR-PCR–based approaches, the theoretical limit for MRD detection by CD34+ DC analysis corresponds to 0.001% to 0.005%.12 This level of sensitivity complies with ELN guidelines for MRD detection in AML, but is still below maximum sensitivities achieved by qPCR.4
To further improve MRD detection in patients with AML lacking a suitable marker for qPCR-based quantification, we established a novel approach, which combines sorting of CD34+ cells from PB with a targeted, error-reduced deep-sequencing procedure (sensitivity of 0.1% to 0.01%) for the sensitive detection of leukemia-specific mutations as markers for residual disease. To evaluate the applicability of our combined approach for routine clinical MRD assessment, we retrospectively analyzed 429 PB and 55 BM samples of 40 patients with AML or MDS in CR after allogeneic HSCT. We demonstrate that the NGS-based detection of MRD in sorted CD34+ (and CD117+) cells is feasible and shows superior sensitivity and specificity compared with CD34+ DC-based analysis. This allows for MRD detection at levels down to 1:106, and detects MRD substantially earlier (median of ∼7 weeks), as compared with conventional CD34+ DC or to the use of genomic DNA from unsorted samples as templates for NGS. In addition, we document the applicability of our approach for panel sequencing in sorted CD34+ PB cells for the early assessment of clonal trajectories in hematological CR.
Methods
Patients
Patients were ≥18 years, had MDS or AML according to World Health Organization criteria, and were enrolled in the RELAZA2 open-label, multicenter, phase 2 trial. The RELAZA2 trial investigated MRD-triggered preemptive treatment with AZA to prevent hematological relapse in patients with MDS or AML (registered on www.clinicaltrials.gov as NCT01462578). For the original study protocol see Platzbecker et al.14 All samples and clinical data were obtained with the written informed consent of the patients. All studies involving human primary material were performed after approval of the local ethics board at each of the participating institutions and were in agreement with the Declaration of Helsinki. In the present study, 29 patients (2 MDS and 27 AML) with molecular relapse (based on a decrease of CD34+ or CD117+ DC <80%) in hematological CR after allogeneic HSCT were retrospectively analyzed for MRD based on NGS (Figure 1). In addition, 11 patients with AML without molecular relapse were included as MRD-negative control subjects. All 40 patients (23 male and 17 female; median age, 62 years; range, 24-75), had documented CD34 (n = 35) or CD117 (n = 5) positivity (>10% of leukemic blasts) by flow cytometry at diagnosis, available sequential CD34+ or CD117+ donor chimerism data, and a known leukemia-specific molecular marker. Time to detection of MRD was compared between different methods and biomaterial templates (PB and BM) in matched serial follow-up samples (429 PB and 55 BM). Based on existing data, a decrease of CD34+ or CD117+ DC <80% was used as the gold standard for MRD positivity (Figure 1A).11,13 In comparison, an error-reduced NGS-based approach (as described later, in “Error-reduced targeted deep sequencing”) was used to monitor MRD in sorted CD34+or CD117+ cells (Figure 1B) as well as in genomic DNA of unsorted material (whole blood; Figure 1C). Throughout the manuscript, the sorted cell populations will be referred to as "CD34+ /CD117+", which means that these cells were either CD34+ or CD117+.
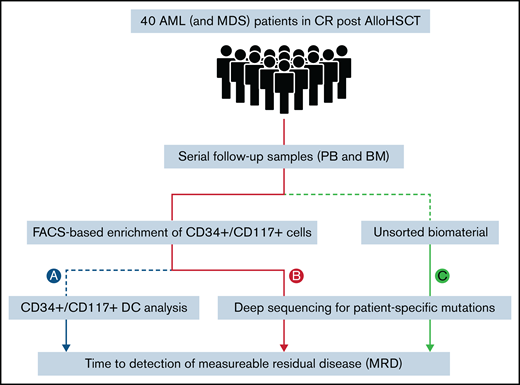
Study design. Processing of patient samples (PB and BM) for MRD detection using different methods: CD34+/CD117+ DC analysis (A); and targeted error-reduced deep sequencing of known leukemia-specific molecular lesions in sorted CD34+/CD117+ cells (B); or in unsorted material (whole blood) (C).
Study design. Processing of patient samples (PB and BM) for MRD detection using different methods: CD34+/CD117+ DC analysis (A); and targeted error-reduced deep sequencing of known leukemia-specific molecular lesions in sorted CD34+/CD117+ cells (B); or in unsorted material (whole blood) (C).
Magnetic cell separation and FACS
To ensure consistent enrichment of CD34+/CD117+ cell populations to high purity (ie, >90%), CD34+/CD117+ cells were enriched by using a combination of magnetic cell separation (MACS) and FACS for all samples. Mononuclear cells (MNCs) were extracted by density gradient centrifugation (500g; 20 minutes; Lymphoprep; Pharmacia, Freiburg, Germany) from 40 to 50 mL of heparinized PB (or 2 mL BM), by using 15 mL Biocoll Separating Solution (Biochrom AG, Berlin, Germany). After centrifugation, the interphase with the MNCs was carefully removed, washed in 10 mL phosphate-buffered saline, and centrifuged for another 5 minutes (500g). The cell pellet was recovered and washed in 10 mL phosphate-buffered saline twice. CD34+/CD117+ cells were isolated from MNCs using MACS by positive selection with the CD34+ or CD117+ Microbead Kit (Miltenyi Biotec, Bergisch-Gladbach, Germany), according to the manufacturer’s recommendations. For FACS-sorting of CD34+/CD117+ cells, the CD34- or CD117-enriched fractions were incubated (15 minutes) with the monoclonal antibodies CD45 FITC/CD34 PE (341071; BD Biosciences, San Jose, CA). Sorting of the CD34+/CD117+ cells was then conducted on a BD FACS Aria II cell sorter (BD Biosciences), aiming for 5 000 to 10 000 CD34+/CD117+ cells and a purity of >90%.
DNA extraction
For DNA extraction, the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) or the ZR Viral DNA Kit (Zymo Research, Orange, CA) (for CD34+/CD117+ cell counts <1000) was used, according to the manufacturers’ protocols. DNA was eluted in 30 to 100 µL of Tris/EDTA buffer.
Error-reduced targeted deep sequencing
Sequencing of genomic DNA from sorted CD34+/CD117+ cells and unsorted material was performed according to an optimized protocol for error-reduced NGS-based detection of low-level, single-nucleotide variants, as described previously.15 In brief, fusion PCR primers for the preparation of amplicon libraries were designed (Primer Premier 6; Premier Biosoft, Palo Alto, CA) according to the manufacturer’s recommendations (Fusion Method; Life Technologies). PCR on genomic DNA of whole blood and sorted CD34+ cells (40 cycles) was performed with Q5 High-Fidelity proofreading polymerases (New England Biolabs, Beverly, MA). PCR primer sequences and specific PCR conditions for all target regions are listed in supplemental Table 1. After Fusion PCR on a GeneAmp PCR System 9700 (Applied Biosystems, Foster City, CA), purified and barcoded PCR products were diluted to 30 pM and loaded on an Ion Chef instrument (Life Technologies) for automated template preparation. Deep sequencing (aiming for >100 000-fold coverage) was conducted on an Ion S5 XL NGS system (Life Technologies). Raw data alignment of demultiplexed FastQ files, variant calling, and filtering were performed with the Sequence Pilot software package (JSI Medical Systems GmbH, Ettenheim, Germany) with default settings. Human genome build HG19 (http://genome.ucsc.edu/) was used as the reference genome for mapping algorithms. Based on mutation-specific false-positive error rates, detection of molecular MRD was conducted with a sensitivity of 0.01% VAF for all targets analyzed (supplemental Figure 1). VAFs below the predefined threshold were considered MRD negative.
NGS panel sequencing of genomic DNA and sorted CD34+ cells
Mutational profiles for the selection of a suitable molecular MRD marker for deep sequencing of CD34+ cells were analyzed in material at diagnosis (50 ng of genomic DNA [gDNA] from whole PB), using the Archer VariantPlex Myeloid panel (ArcherDX, Boulder, CO), which covers 75 genes (and mutational hotspots) frequently associated with myeloid neoplasms, according to the manufacturer’s instructions. In addition, DNA from sorted CD34+ PB cells from selected patients after allo-HSCT was processed with the VariantPlex Myeloid panel (ArcherDX) to assess potential molecular shifts and clonal trajectories in hematological CR. Eligible samples used for panel sequencing of sorted CD34+ cells had a CD34+ DC <80% and contained 1500 to 5000 cells (allowing for the extraction of 9-30 ng DNA). Libraries were sequenced on a MiSeq (300-bp paired end) NGS instrument (Illumina, San Diego, CA) with a median coverage of 1050 reads per target region (range, 170-2830). Mutation calling was performed with the Archer Analysis Software Package (version, 6.2.7), with a defined cutoff of 5% VAF.
Dilution experiment
A blood sample (200 mL) was drawn from a healthy volunteer donor. Sampling was repeated 2 times to achieve a total of 3 replicate dilution series. The cell count was 6.77 × 106 to 1.24 × 107 WBCs per milliliter. The concentration of CD34+ cells in this sample was 0.08%. For each dilution series, 5 aliquots were prepared, each containing 30 mL of whole blood. To each aliquot, cells of a CD34+ AML-cell line (Kasumi; DSMZ, Braunschweig, Germany) carrying the KIT N822K single-nucleotide variant were added to obtain dilutions of 1:10 000, 1:50 000, 1:100 000, 1:500 000, and 1:1 000 000 Kasumi cells in normal WBCs. 20 mL of each aliquot was then processed for the FACS-based enrichment of CD34+ cells and subsequent NGS for the detection of the KIT N822K variant (as described in “Correlation between methods for MRD assessment”). In addition, multiplex STR-PCR for CD34+ DC analysis was performed, as described previously.16
Ethics approval
All samples were collected with written informed consent and after the local ethics board at each of the participating institutions approved the study.
Statistical analysis
Categorical variables between groups were compared by 2-sided Student t test. For continuous variables, the nonparametric Mann-Whitney U test was applied. The nonparametric Spearman test was performed to calculate correlations between different MRD detection methods. Calculations were conducted in Prism 5 (GraphPad, La Jolla, CA). P < .05 was considered significant. Sensitivity and specificity to predict relapse were calculated by the R Environment for Statistical Computing, version 4.0.3. The threshold maximizing the Youden-Index, area under the curve, and accuracy were estimated as mean values derived from 10-fold cross-validated receiver operating characteristic analyses.
Results
Deep sequencing in sorted CD34+/CD117+ cells
For all samples processed, CD34+/CD117+ cell counts obtained by FACS (median 4200; range 70-8700) (supplemental Figure 2A) allowed for collection of sufficient DNA (median, 18.5 ng; range, 1.2-72.3) for PCR amplification, and subsequent targeted deep sequencing, with a median coverage of 96 385 reads (range, 13 241-449 401). Generally, there was a moderate trend (Spearman r = −0.3405) for higher CD34+/CD117+ cell counts in samples with lower CD34+/CD117+ DC values (supplemental Figure 2B), probably because of the presence of an increased number of circulating CD34+/CD117+ AML cells in the PB in patients with imminent relapse. For retrospective genetic MRD testing of clinical samples, 1 somatic AML-specific mutation was selected per patient (supplemental Table 1). For monitoring, clonal (dominant) mutations with a VAF of >20%, which likely represent early molecular events that are present in the CD34+ AML cell fraction, were preferred for analysis. Most common genetic alterations used as MRD markers were mutations in TP53 (n = 6), RUNX1 (n = 6) and U2AF1 (n = 4) genes (supplemental Figure 3). Whenever possible, potential CHIP variants in DTA genes (DNMT3A, TET2, ASXL1)17 were excluded for MRD detection of clinical samples. In 3 patients (3, 26, and 27) without other eligible MRD markers, mutations of TET2 were used as molecular markers. However, although the selection of non-DTA mutations may be relevant for genetic MRD testing in patients with other treatments (ie, chemotherapy), the detection of patient-specific DTA mutations after allogeneic hematopoietic HSCT most likely indicates the presence of residual recipient cell populations and MRD, respectively.
Correlation between methods for MRD assessment
To assess the comparability between all different MRD methods tested, values were correlated between NGS and chimerism-based analysis of matched patient samples. A strong correlation (Spearman r = −0.8601; P < .0001) of MRD levels was observed between the STR-PCR (CD34+ DC) and NGS-based analysis of sorted CD34+/CD117+ PB cells (Figure 2A). Accordingly, CD34+/CD117+ DC values <80% were associated with an NGS-based MRD-positivity rate (>0.01% VAF) of 97% to 100% in CD34+/CD117+ PB cells, which demonstrates robust and largely congruent MRD detection in both assays (Figure 2B). Moreover, the NGS-based method performed on sorted CD34+/CD117+ PB cells detected MRD in 54% and 24% in samples considered MRD-negative based on CD34+/CD117+ DC values of >95% and >99%, respectively (Figure 2B). Likewise, the detection of the CD34+ Kasumi cell line in peripheral WBCs was feasible up to a dilution of 1:105, using STR-PCR (CD34+ DC mean, 93.83% ± 2.04%) on sorted CD34+ cells, but increased ∼10-fold (up to 1:106) by the application of NGS for the quantification of the KIT N822K mutation in CD34+ cells (Kasumi cell/WBC dilution 1:1 000 000; NGS CD34+ cells VAF 0.42%; CD34+ DC >99.9%; Figure 2C). Although there was a strong correlation (Spearman r = 0.8991; P < .0001) between the use of PB and BM as a template for NGS- and DC-based analyses (Figure 2D; supplemental Figure 4, respectively), sensitivity for MRD detection in sorted CD34+ cells was increased by the use of PB, when compared with BM aspirates, for initial FACS enrichment. For example, median VAFs detected in sorted CD34+ cells were substantially higher in PB (8.75%), compared with those detected in matched BM aspirates (0.91%), although the difference was not significant; P =.1377; Figure 2E). Furthermore, a positive correlation (Spearman r = 0.6202; P < .0001; supplemental Figure 5) was observed for the comparison of allelic ratios obtained by NGS of sorted CD34+/CD117+ cells (median, 1.27; interquartile range, 0.003% to 20.19% VAF) vs the use of gDNA from unsorted whole blood (median, 0.0046; interquartile range, 0.001% to 0.039% VAF; P < .0001) as a PCR template, with significantly higher (∼1000-fold) VAFs detected in CD34+ PB cells (Figure 2F).
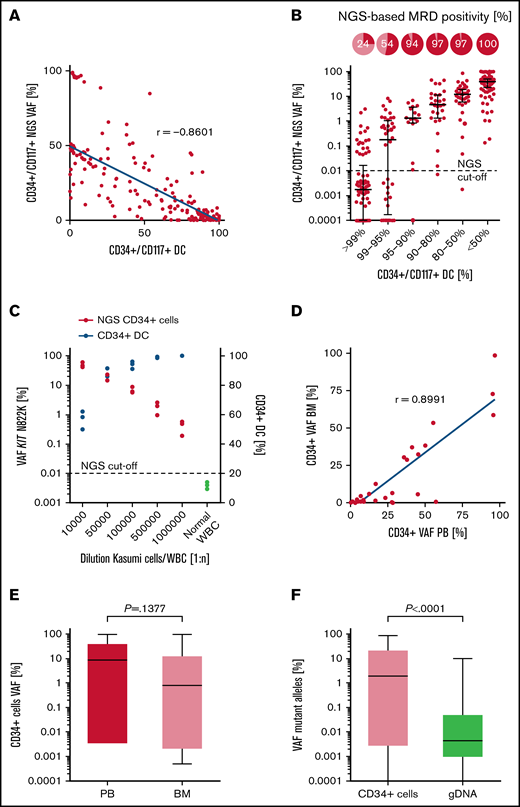
Technical evaluation of NGS-based MRD detection in CD34+/CD117+ cells. (A) Correlation of MRD detection using NGS or DC analysis in sorted CD34+/CD117+ PB cell samples (n = 267). (B) NGS-based MRD positivity rates in relation to the corresponding CD34+/CD117+ DC level in PB. The cutoff for NGS-based MRD quantification is indicated at 0.01% VAF. (C) Detection of the Kasumi cell line in PB using NGS-based quantification of the KIT N822K variant (red dots) or by CD34+ DC analysis (blue dots). (D) Correlation of NGS-based MRD detection in sorted CD34+ cells of matched PB and BM samples (n = 35) as templates for analysis. (E) Quantification of variant allele frequencies (%) in CD34+ cells using matched PB or BM samples (n = 35) for NGS. (F) Quantification of mutant alleles by NGS using sorted CD34+/CD117+ PB cells or unsorted material of matched follow-up samples (n = 197). Box plots represent median values with interquartile range; box whiskers represent minimum to maximum values.
Technical evaluation of NGS-based MRD detection in CD34+/CD117+ cells. (A) Correlation of MRD detection using NGS or DC analysis in sorted CD34+/CD117+ PB cell samples (n = 267). (B) NGS-based MRD positivity rates in relation to the corresponding CD34+/CD117+ DC level in PB. The cutoff for NGS-based MRD quantification is indicated at 0.01% VAF. (C) Detection of the Kasumi cell line in PB using NGS-based quantification of the KIT N822K variant (red dots) or by CD34+ DC analysis (blue dots). (D) Correlation of NGS-based MRD detection in sorted CD34+ cells of matched PB and BM samples (n = 35) as templates for analysis. (E) Quantification of variant allele frequencies (%) in CD34+ cells using matched PB or BM samples (n = 35) for NGS. (F) Quantification of mutant alleles by NGS using sorted CD34+/CD117+ PB cells or unsorted material of matched follow-up samples (n = 197). Box plots represent median values with interquartile range; box whiskers represent minimum to maximum values.
Feasibility for early MRD detection in the clinical setting
To address the feasibility of our combined approach for early MRD assessment in the clinical setting, stored material from 40 patients with a median follow-up of 818 days (range, 101-2263) after allogeneic HSCT was retrospectively analyzed for the detection of mutant alleles in sorted CD34+/CD117+ cells from PB (and gDNA from whole blood; Figure 3A; supplemental Figures 6 and 7). Based on a decrease of CD34+/CD117+ DC <80%, molecular relapse occurred after a median of 210 days (range, 94-706) after allo-HSCT (Figure 3B). Patients with confirmed MRD positivity in CR received treatment with AZA (75 mg/m2 per day for up to 24 cycles) until hematological relapse (ie, patient 10) or until major response (MRD negativity; ie, patient 1) (Figure 3A). In all patients analyzed, molecular relapse was associated with an increase of respective mutant alleles in sorted CD34+/CD117+ cells before (in 86.2% of patients; patients 1, 2, 4, 5, 7-18, 20-23, and 25-29) or simultaneous with CD34+/CD117+ DC analysis (in 4 patients; 3, 6, 19, and 24; Figure 3B; supplemental Figure 6). In addition, NGS-based analysis of CD34+/CD117+ PB cells allowed for MRD detection significantly earlier than with the use of gDNA from unsorted whole blood for NGS (patients 1, 3-5, 8, 10, 11, 13-16, 18, 21-23, and 25-29) in most of the patients (Figure 3B; supplemental Figure 6). Interestingly, in 2 patients (6 and 9) with early recurrence (151 and 109 days, respectively), NGS-based MRD detection in unsorted blood was feasible before the use of matched CD34+ cell samples, with sufficient cell counts for analysis (Figure 3B; supplemental Figure 6). In line with this finding, the diagnostic advantage of NGS-based MRD detection in CD34+/CD117+ PB cells (compared with whole blood) was generally less pronounced in patients with early molecular relapse (Figure 3B), presumably because of the faster growth kinetics of the leukemic clone in those patients. No significant differences in the diagnostic performance were observed between patients with AML and the 2 patients with high-risk MDS or between the use of DTA or non-DTA mutations as MRD markers. Also, no differences were seen with the use of CD34+ or CD117+ cells as templates for NGS, although the number of CD117+ patients was small (n = 5). However, in general, the NGS-based analysis of sorted CD34+/CD117+ cells allowed for MRD detection significantly earlier than the use of CD34+/CD117+ DC (median, 48 days; range, 0-281; P = .0011) or NGS of whole-blood samples (median, 93 days; range, 0-1141; P < .001) (Figure 3C). Notably, during administration of AZA, the kinetics of mutant alleles detected in CD34+/CD117+ cells reflected the treatment response (ie, patients 1, 2, 12, 16, and 26; Figure 3A; supplemental Figure 6), which may also indicate the potential applicability of our combined approach for precise therapy monitoring. Similarly, preliminary results of a prospective cohort indicate that the NGS-based analysis of AML-specific mutations in sorted CD34+ cells enables monitoring of treatment efficiency and MRD, also in patients with AML treated with chemotherapy, with sensitivities superior to conventional MFC and similar to qPCR-based NPM1 mutation-specific assays (supplemental Figure 8).
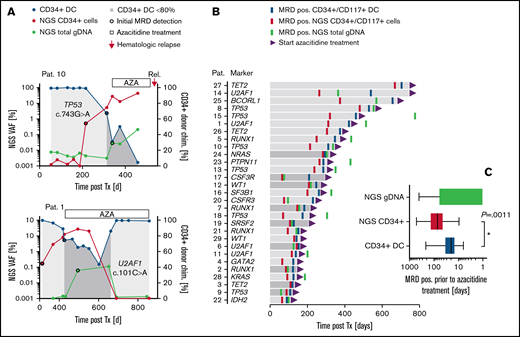
MRD detection in clinical samples after allogeneic HSCT. (A) Representative MRD detection in 2 patients with (patient 1) or without (patient 10) major response post AZA treatment. For MRD quantification different methods were applied: CD34+ DC-based analysis (blue dots); NGS on FACS-enriched CD34+ PB cells (red dots) or whole-blood samples (green dots). For CD34+ DC analysis, values <80% were considered MRD positive. For NGS-based analysis, an increase in mutant alleles >0.01% was classified MRD positive. (B) Follow-up intervals after allo-HSCT and MRD detection (coded by color) in 29 patients with MRD-guided preemptive AZA treatment. An improvement of MRD detection by NGS-based analysis of CD34+/CD117+ PB cells (as compared with both other MRD methods tested) is indicated by the light gray bars. (C) MRD detection before AZA treatment. Box plots represent median values with interquartile range; box whiskers represent minimum to maximum values.
MRD detection in clinical samples after allogeneic HSCT. (A) Representative MRD detection in 2 patients with (patient 1) or without (patient 10) major response post AZA treatment. For MRD quantification different methods were applied: CD34+ DC-based analysis (blue dots); NGS on FACS-enriched CD34+ PB cells (red dots) or whole-blood samples (green dots). For CD34+ DC analysis, values <80% were considered MRD positive. For NGS-based analysis, an increase in mutant alleles >0.01% was classified MRD positive. (B) Follow-up intervals after allo-HSCT and MRD detection (coded by color) in 29 patients with MRD-guided preemptive AZA treatment. An improvement of MRD detection by NGS-based analysis of CD34+/CD117+ PB cells (as compared with both other MRD methods tested) is indicated by the light gray bars. (C) MRD detection before AZA treatment. Box plots represent median values with interquartile range; box whiskers represent minimum to maximum values.
Diagnostic performance for relapse prediction
To evaluate the diagnostic performance of our approach in more detail, 11 patients with AML without molecular relapse were analyzed for the detection of MRD (supplemental Figure 7). Leukemia-specific VAFs in sorted CD34+ cells were below the predefined technical detection limit of 0.01% throughout the entire course of follow-up in 9 of 11 patients. In 2 patients (34 and 36), a transient increase of VAFs up to 0.25% and 0.03%, respectively, was not associated with subsequent relapse. Hence, when applying a threshold of 0.01% for NGS-based MRD detection in CD34+ cells, relapse prediction would be possible with a sensitivity of 100% and specificity of 81.8%. To optimize the accuracy of our test, receiver operating characteristic curve analysis on data of all 40 patients reported an optimal NGS cutoff of 0.0385% (with superior area under the curve of 0.996) for confident identification of patients with imminent relapse with superior sensitivity (100%) and specificity (91%) (supplemental Figure 9).
Assessment of clonal trajectories in complete remission
The assessment of MRD via the NGS-based detection of a leukemia-specific mutation in sorted CD34+ cells was possible in 100% of patients with confirmed molecular relapse (n = 19). However, in patient 3, a FLT3TKD D835Y mutation, identified at AML diagnosis, was not detected in MRD-positive (based on CD34+ DC analysis) follow-up samples after allo-HSCT (Figure 4A), pointing at clonal selection in hematological CR. In contrast, a TET2 insertion variant of the same patient (used as alternative MRD marker) increased in association with the decrease of CD34+ DC values after allo-HSCT (Figure 4A). To address potential clonal evolution during CR in more detail, we performed NGS panel analysis in sorted CD34+ PB cells, which confirmed the loss of the FLT3TKD D835Y variant (as well as the loss of another lesion in FLT3 and CBL) at day 131 after allo-HSCT, whereas 2 novel mutations (in EZH2 and TET2) emerged in hematological CR (Figure 4B-C). After partial response during AZA treatment (Figure 4A), the patient acquired another 3 mutations (2 x BCORL1 and GATA2) detected at molecular relapse (day 455 after allo-HSCT; Figure 4B), which was followed by hematological relapse at day 505 after allo-HSCT (Figure 4A,C). Similar shifts of leukemic clones in hematological CR after allo-HSCT and during administration of AZA were detected in sorted CD34+ PB cells of 2 other patients (ie, patient 11, gain of lesions in ASXL1 and RUNX1; and 14, gain of lesions in BCOR, CUX1, IKZF1, RUNX1, and NRAS; Figure 4D).

Early assessment of clonal trajectories in CD34+ cells. (A) MRD level after allo-HSCT in a patient with temporary response to AZA treatment. For NGS-based MRD assessment, 2 available molecular marker (FLT3 c.2503G>T and TET1 c.5717insAATAG) were used. (B) Results of the NGS panel analysis in sorted CD34+ cells of MRD-positive samples obtained 131 and 455 days after allo-HSCT. (C) Clonal trajectories depicted from molecular profiles at AML diagnosis and CD34+ cells in complete hematological remission. (D) Detection of molecular lesions and corresponding variant allele frequencies (%) at AML diagnosis (gDNA from unsorted whole blood) and in CD34+ PB cells, sampled at the time of molecular relapse (after allo-HSCT) and hematological relapse (after AZA treatment) in 2 patients.
Early assessment of clonal trajectories in CD34+ cells. (A) MRD level after allo-HSCT in a patient with temporary response to AZA treatment. For NGS-based MRD assessment, 2 available molecular marker (FLT3 c.2503G>T and TET1 c.5717insAATAG) were used. (B) Results of the NGS panel analysis in sorted CD34+ cells of MRD-positive samples obtained 131 and 455 days after allo-HSCT. (C) Clonal trajectories depicted from molecular profiles at AML diagnosis and CD34+ cells in complete hematological remission. (D) Detection of molecular lesions and corresponding variant allele frequencies (%) at AML diagnosis (gDNA from unsorted whole blood) and in CD34+ PB cells, sampled at the time of molecular relapse (after allo-HSCT) and hematological relapse (after AZA treatment) in 2 patients.
Discussion
In this study, we evaluated and confirmed the feasibility of a new NGS-based method for sensitive MRD monitoring in CD34+ PB cells of patients with CD34+ AML or high-risk MDS. In addition, we documented the applicability and superior sensitivity of this approach for early prediction of disease recurrence in the clinical setting.
Several studies reported an advantage of the complementary use of >1 MRD detection technique (ie, the parallel application of MFC and qPCR) for the prediction of relapse in high-risk myeloid malignancies.17-20 In contrast to these reports, our 2-step approach highly improved sensitivity for MRD detection by the combination of initial FACS-based enrichment of circulating CD34 cells in PB (typically encompassing 0.01% to 0.1% of WBCs in PB) and subsequent targeted error-reduced deep sequencing (with a sensitivity of 0.01% VAF). Consequently, quantification of AML-specific mutations in CD34+ PB cells increased by a factor of ∼1000, compared with the use of DNA from unsorted whole PB for NGS, down to levels of 10−5 to 10−6. This level of sensitivity is comparable to or even superior to qPCR-based approaches.7,8 Unlike qPCR, our NGS procedure does not rely on the presence of specific fusion genes or mutations such as NPM1 and is hence applicable to most patients with AML with a detectable molecular marker. Moreover, although our approach was applied to patients with CD34 antigen expression at diagnosis in the present study, FACS-based enrichment can easily be adapted to other patient-specific immunophenotypes. In those few patients lacking CD34+ expression on their leukemic blasts, CD117+ cells were successfully used, further expanding the group of patients who are candidates for this approach. Other groups recently presented similar strategies, (ie, a recent study showed an advantage of using a whole combinatory antibody panel for MFC-based leukemic cell enrichment followed by mutational analysis to analyze most of the patients with AML and to cover potential immunophenotypic shifts during MRD monitoring21 ). However, previous studies on immunophenotypic differences between diagnosis and relapse in AML reported a predominance of more immature phenotypes at relapse, with an increase of the expression of CD34 and/or CD117 in the relapse samples, whereas the loss of these early antigens is reported only occasionally (ie, in 4% of patients).22 Using conventional NGS (sensitivity of 0.1% to 0.5% VAF), the latter work21 reported the detection of 1 leukemic cell in 10 000 normal BM cells. In contrast, our error-reduced NGS procedure enabled the detection of 1 Kasumi cell in 1 million WBCs in PB. When compared with the use of BM aspirates, the use of PB is acknowledged as an alternative source for minimally invasive MRD assessment in the current ELN recommendations.4,23 The potential use of PB for the reproduction of BM molecular profiles was previously demonstrated by the application of NGS- or fluorescence in situ hybridization–based analysis on immunomagnetic enriched CD34+ cells from PB.24,25 In line with previous observations,11 our data indicate that the sensitivity for MRD detection (using both DC and NGS) in CD34+-sorted PB samples is at least equal to or even higher than that in CD34+ cells from BM aspirates. This finding may indicate that a higher proportion of malignant CD34+ cells (vs normal CD34+ cells) are released into PB circulation from the BM environment, and/or a delayed clearance of leukemic blasts in peripheral circulation occurs after treatment. Most important, we demonstrate that the detection of molecular relapse is feasible and identifies patients at risk of relapse a further 7 weeks before the CD34+ DC assessment, which potentially provides an even longer interval for therapeutic intervention (ie, with AZA or novel drugs; eg, venetoclax or immunotherapeutic approaches).8 Although only 2 cases of high-risk MDS were included in the present cohort of patients with AML, disease dynamics and early MRD detection are likely to vary between these entities (ie, shorter interval between MRD detection and relapse in AML), which has to be considered in the clinical setting. Currently, a major drawback of our approach for frequent monitoring is the high volume of blood (40-50 mL) typically needed to obtain a sufficient number of CD34+ cells for targeted deep sequencing. However, initial data using a novel approach for cell enrichment, based on hydroacoustic focusing and subsequent CD34+ enrichment (MARS-SP/MARS-BAR system; Applied Cells, Palo Alto, CA) indicate that the necessary blood volumes may be reduced substantially (down to 10-15 mL). Furthermore, as compared with CD34+ DC (which relies on the detection of donor/recipient cell signals), our method is also applicable to patients with AML without allogeneic HSCT by the direct quantification of leukemia cells in PB. Therefore, molecular markers used for MRD assessment need careful evaluation, as mutations in DTA genes may also occur during normal age-related CHIP and hence may not necessarily represent the presence of MRD.4,17,26 Likewise, in patients undergoing allo-HSCT, recurrent DTA hotspot mutations (ie, R882 DNMT3A) frequently associated with CHIP should be tested in both recipient and donor material before their use as molecular MRD markers, to exclude false-positive MRD results related to the detection of donor CHIP. Also, mutations of the RAS signaling pathway may be frequently lost or emerge during the course of disease (ie, FLT3, CBL, and NRAS in our study), as consequence of treatment related clonal selection and with relevance for postremission strategies.27 In this regard, we demonstrate the applicability of our approach for panel sequencing in sorted CD34+ cells from PB obtained in hematological CR, which potentially allows for the early assessment of novel emerging leukemia subclones and the identification of new drug targets, respectively. Recently, a similar approach based on FACS-based preenrichment of leukemic immunophenotypes and subsequent NGS panel analysis, enabled the detection of subclonal shifts and the assessment of clonal hierarchy in chronic lymphocytic leukemia and patients with AML.28,29 However, as a practical constraint for longitudinal MRD monitoring, NGS panel sequencing increases analysis cost per sample 5- to 10-fold at a lower sensitivity (ie, 2% to 5% VAF detection cutoff in the sorted population with an overall sensitivity of ∼1:50 000 in total WBCs), compared with the use of a single molecular marker for deep sequencing (reagent costs of ∼50 € in our study, with a detection limit of 0.01% in the sorted cells, ie, overall sensitivity of ∼1:1 000 000 in total WBCs). Therefore, instead of using whole-gene panels, targeted deep sequencing of multiple genomic loci (ie, 2-3 patient-specific mutations) in CD34+ cells could provide greater sensitivity for MRD detection in clinical routine.4 This finding may become especially important, in the light of the many new targeted agents currently becoming available (eg, IDH1/2 inhibitors,30 CD47 antibodies,31 and others). In addition, in patients with negative MRD, immunosuppressive therapy can be safely extended to minimize the risk of GvHD. In our feasibility study, we retrospectively investigated a preselected cohort of patients with AML and patients with high-risk MDS with known MRD status. Therefore, results are currently validated, in prospective evaluations with larger samples of patients.
In summary, we propose a novel, easily accessible, robust method for ultrasensitive MRD detection in PB that is applicable to most patients with AML. Initial results demonstrated the feasibility of targeted deep sequencing of CD34+ cells for early relapse prediction in clinical settings, with superior sensitivity and specificity, as compared with chimerism-based MRD assessment or the use of unsorted PB for NGS.
Acknowledgments
The authors thank M. Böhm, M. Hartwig, and J. Bornhäuser for skilled technical assistance.
Authorship
Contribution: C.T., S.S., and C.B.-M. conceived of the work; C.T., C.B.-M., S.S., J.M.M., H.S., C.M.-T., C.D.B., K.S., C.R., and U.P. collected samples and data; C.B.M., S.S., and U.O. acquired and analyzed the data; S.S. and C.B.-M. performed the bioinformatics analysis; M.K. performed the statistical analysis; C.T., C.B.-M., and S.S. interpreted the data; S.S. and C.T. drafted the manuscript; G.E., M.B., and U.P. provided administrative support; and all authors reviewed and approved the manuscript for submission.
Conflict-of-interest disclosure: C.T. is CEO and co-owner of AgenDix GmbH, a company that performs molecular diagnostics. The remaining authors declare no competing financial interests.
Correspondence: Christian Thiede, Universitätsklinikum Carl Gustav Carus, Medizinische Klinik und Poliklinik I, Fetscherstraße 74, 01307 Dresden, Germany; e-mail: christian.thiede@uniklinikum-dresden.de.

