

Key Points
Loss of G6b-B leads to an unexpected megakaryocyte development defect resulting in severe macrothrombocytopenia.
G6b-B–deficient mice display reduced levels of MK-specific transcripts, surface receptors, GATA-1, and thrombopoietin signaling.
Abstract
G6b-B is a megakaryocyte lineage-specific immunoreceptor tyrosine-based inhibition motif–containing receptor, essential for platelet homeostasis. Mice with a genomic deletion of the entire Mpig6b locus develop severe macrothrombocytopenia and myelofibrosis, which is reflected in humans with null mutations in MPIG6B. The current model proposes that megakaryocytes lacking G6b-B develop normally, whereas proplatelet release is hampered, but the underlying molecular mechanism remains unclear. We report on a spontaneous recessive single nucleotide mutation in C57BL/6 mice, localized within the intronic region of the Mpig6b locus that abolishes G6b-B expression and reproduces macrothrombocytopenia, myelofibrosis, and osteosclerosis. As the mutation is based on a single-nucleotide exchange, Mpig6bmut mice represent an ideal model to study the role of G6b-B. Megakaryocytes from these mice were smaller, displayed a less-developed demarcation membrane system, and had a reduced expression of receptors. RNA sequencing revealed a striking global reduction in the level of megakaryocyte-specific transcripts, in conjunction with decreased protein levels of the transcription factor GATA-1 and impaired thrombopoietin signaling. The reduced number of mature MKs in the bone marrow was corroborated on a newly developed Mpig6b-null mouse strain. Our findings highlight an unexpected essential role of G6b-B in the early differentiation within the megakaryocytic lineage.
Introduction
Megakaryocytes (MKs) are large, polyploid cells within the bone marrow (BM) that develop in a complex differentiation process. Mature MKs harbor granules and internal membrane systems, which are indispensable for the assembly and release of functional platelets.1 G6b-B has been identified as an essential regulator of platelet biogenesis.2 Deletion of the Mpig6b locus in mice results in macrothrombocytopenia and myelofibrosis,2 which was recently recapitulated in humans with disease-causing null variants within MPIG6B.3-7 Based on unaltered ploidy and thrombopoietin (TPO)-induced Erk1/2 activation, reduced proplatelet formation, and spreading of Mpig6b−/− MKs in vitro, it has been hypothesized that G6b-B does not play a role in development of MKs, but in a terminal step in platelet production.2,8 However, the underlying molecular mechanism of how this receptor regulates thrombopoiesis has remained unknown. By characterizing a spontaneous Mpig6b mutant together with a newly generated Mpig6b-null mouse line we provide unexpected experimental evidence that establishes G6b-B as a central regulator of transcription during MK maturation, which can explain the complex phenotype of these mice.
Methods
Homozygous mutant mice carrying a spontaneously developed mutation in a splice acceptor site of Mpig6b (NM_001033221.3; c.404-1G>A) are referred to as Mpig6bmut. Platelet and MK isolation, whole-exome sequencing, RNA sequencing, transmission electron microscopy, flow cytometry, ELISA, platelet recovery, tail bleeding, histology, immunofluorescence stainings, MK ploidy, qPCR, immunoblotting, and data analysis are described in the supplemental Methods.
Results and discussion
Single-nucleotide exchange in Mpig6b results in macrothrombocytopenia
In a breeding colony of C57BL/6 mice, we identified individual animals with bleeding related to severe macrothrombocytopenia (Figure 1A-B). Ten generations of backcrossing led to isolation of a substrain presenting with a recessive trait. Using whole-exome sequencing, we identified a homozygous single-nucleotide exchange within Mpig6b (c.404-1G>A) in 10 of 10 mice (allele frequency: 1.0), resulting in abolished G6b-B protein expression in platelets (Figure 1C-E; supplemental Figure 1A-B). In silico prediction unraveled the introduction of a splice acceptor site in intron 2 of Mpig6b, leading to an out-of-frame shift transcript with multiple stop codons, expected to result in nonsense-mediated messenger RNA (mRNA) decay. The absence of G6b-B led to a reduction in surface expression levels of platelet membrane glycoproteins (GPs; supplemental Table 1), infinite tail bleeding times, myelofibrosis, splenomegaly, and additional osteosclerosis in female Mpig6bmut mice (supplemental Figure 1C-F). We thus identified a spontaneous single-nucleotide mutation within Mpig6b, resulting in a phenotype that faithfully recapitulates Mpig6b−/− and Mpig6b diY/F mice.2,8,9

Single-nucleotide mutation within Mpig6b results in severe macrothrombocytopenia and impaired MK maturation. Platelet count (A) and volume (B) in 10-week-old female and male WT and Mpig6bmut mice were assessed with an automated blood cell analyzer. Values are the mean ± SD. Unpaired, 2-tailed Student t test. ***P < .001. (C) Identification of a mutation in a splice acceptor site of Mpig6b in Mpig6bmut mice by whole-exome sequencing. The G>A single-nucleotide exchange in Mpig6b was present in all reads in mutant but not in WT mice. (D-E) Absence of G6b-B was validated in Mpig6bmut platelets by flow cytometry (D) and immunoblot analysis (C-terminal antibody) using enhanced chemoluminescence (E). (F) WT and Mpig6bmut MKs were differentiated in vitro in the presence of TPO and analyzed by brightfield microscopy. Mean MK diameter was determined manually with ImageJ software. At least 30 MKs per culture were analyzed. Values are mean ± SD (n = 3). Unpaired, 2-tailed Student t test. *P < .05. (G) The αIIbβ3+ cell population in whole BM of WT or Mpig6bmut mice was analyzed by flow cytometry. Values are mean ± SD (n = 8). Unpaired, 2-tailed Student t test. ***P < .001. (H) Mean size of native MKs was analyzed ex vivo by flow cytometry. Values are mean ± SD (n = 4). Unpaired, 2-tailed Student t test. ***P < .001. (I) Demarcation membrane system maturation in WT and Mpig6bmut BM MKs was visualized by transmission electron microscopy, constrasted by osmium tetroxide and stained with uranly acetate/lead citrate. Bars represent 3 µm; insets: 1.5 µm. Nuclear (J) and DMS (K) fraction in relation to cell size were quantified manually using ImageJ software. At least 7 MKs per mouse were analyzed. Values are mean ± SD (n = 3). Unpaired, 2-tailed Student t test. *P < .05; ***P < .001.
Single-nucleotide mutation within Mpig6b results in severe macrothrombocytopenia and impaired MK maturation. Platelet count (A) and volume (B) in 10-week-old female and male WT and Mpig6bmut mice were assessed with an automated blood cell analyzer. Values are the mean ± SD. Unpaired, 2-tailed Student t test. ***P < .001. (C) Identification of a mutation in a splice acceptor site of Mpig6b in Mpig6bmut mice by whole-exome sequencing. The G>A single-nucleotide exchange in Mpig6b was present in all reads in mutant but not in WT mice. (D-E) Absence of G6b-B was validated in Mpig6bmut platelets by flow cytometry (D) and immunoblot analysis (C-terminal antibody) using enhanced chemoluminescence (E). (F) WT and Mpig6bmut MKs were differentiated in vitro in the presence of TPO and analyzed by brightfield microscopy. Mean MK diameter was determined manually with ImageJ software. At least 30 MKs per culture were analyzed. Values are mean ± SD (n = 3). Unpaired, 2-tailed Student t test. *P < .05. (G) The αIIbβ3+ cell population in whole BM of WT or Mpig6bmut mice was analyzed by flow cytometry. Values are mean ± SD (n = 8). Unpaired, 2-tailed Student t test. ***P < .001. (H) Mean size of native MKs was analyzed ex vivo by flow cytometry. Values are mean ± SD (n = 4). Unpaired, 2-tailed Student t test. ***P < .001. (I) Demarcation membrane system maturation in WT and Mpig6bmut BM MKs was visualized by transmission electron microscopy, constrasted by osmium tetroxide and stained with uranly acetate/lead citrate. Bars represent 3 µm; insets: 1.5 µm. Nuclear (J) and DMS (K) fraction in relation to cell size were quantified manually using ImageJ software. At least 7 MKs per mouse were analyzed. Values are mean ± SD (n = 3). Unpaired, 2-tailed Student t test. *P < .05; ***P < .001.
Analyzing BM-derived MKs from Mpig6bmut mice differentiated in vitro, we found that the number of proplatelet-forming cells was significantly reduced compared with the wild type (WT) (supplemental Figure 2A-D). The cytoskeleton of proplatelet-forming Mpig6bmut MKs, however, appeared morphologically comparable. Intravital 2-photon microscopy of the cranial BM revealed a high degree of MK fragmentation, together with a low amount of circulating platelets in Mpig6bmut mice (supplemental Movies 1-3).
Impaired maturation of Mpig6bmut MKs
The diameter of in vitro–differentiated Mpig6bmut MKs was significantly smaller (Figure 1F). When native BM cells were analyzed flow cytometrically, we observed an increased percentage of MKs in Mpig6bmut mice, also displaying a smaller size (Figure 1G-H). Transmission electron microscopy of BM MKs in situ revealed severely defective maturation of the demarcation membrane system in Mpig6bmut mice (Figure 1I-K). We also observed increased neutrophil emperipolesis into mutant MKs in situ and in cryosections (supplemental Figure 2E-F). Mpig6bmut mice exhibited elevated TPO plasma levels (supplemental Figure 2G), as expected in response to low platelet counts. Ploidy analysis showed a significant increase in 2n megakaryoblasts, whereas the fractions of 16n and 32n MKs were marginally reduced (supplemental Figure 2H-I), which may be a consequence of higher TPO levels and a skewed differentiation toward the MK lineage in the mutant mice. Interestingly, the percentage of small- and medium-sized MKs was markedly increased, whereas the fraction of large, highly granular side scatterhigh (SSChigh) MKs was severely reduced in the BM of Mpig6bmut mice (Figure 2A-B). The expression levels of prominent GPs correlated positively with MK size in WT animals, in which SSChigh MKs exhibited the highest GP surface abundance (supplemental Figure 3A). Notably, the overall GP surface expression in the entire MK population revealed a marked reduction for all GPs in Mpig6bmut mice (Figure 2C), because of the accumulation of immature MKs that expressed lower levels of the respective GPs. These findings were recapitulated in in vitro–differentiated MKs (supplemental Figure 3B-C). We cannot completely rule out a role for the established G6b-B ligand perlecan10 (or other potential ligands) in our culture conditions. The reduced surface expression of GPIbα, α2 integrin, and GPVI agrees with data from Mpig6b−/− and Mpig6bfl/fl;Pf4-Cre+ mice2,11 ; however, the reduction was attributed to shedding of these receptors.2 Interestingly, our finding resembles other mouse lines, where reduced MK maturation is associated with normal ploidy, including Gfi1b−/−,12 Nfe2−/−,13 or Rhoafl/fl;Cdc42fl/fl;Pf4-Cre+14 mice. The accumulation of immature MKs can lead to myelofibrosis, osteosclerosis, or both, as reported for the Gata1low or Nfe2−/− mouse lines15-17 and may explain the phenotypes in G6b-B–null mice.
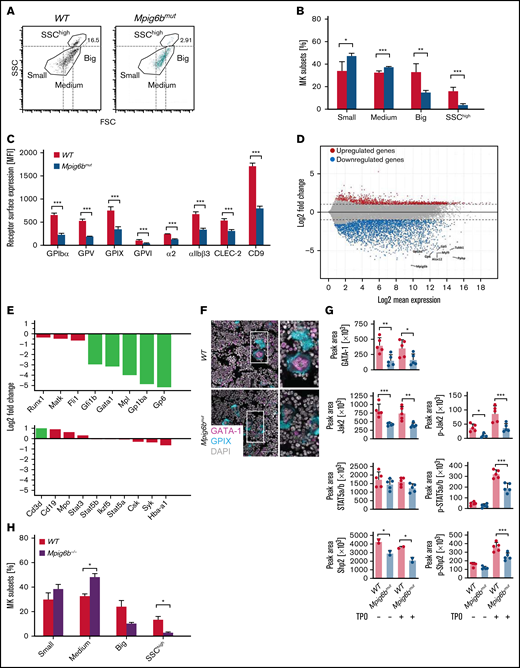
Maturation block in Mpig6bmut MKs involves reduced gene expression and TPO signaling. (A-B) Mean size distribution of WT and Mpig6bmut MKs was analyzed by flow cytometry. (A) Dot plots depicting the proportion of SSChigh MKs and the delineation between small, medium, and large MKs. (B) Values are mean ± SD (n = 6). Unpaired, 2-tailed Student t test. *P < .05; **P < .01; ***P < .001. (C) Mean surface receptor expression on the whole MK population derived from WT and Mpig6bmut mice. Values are mean ± SD (n = 4). Unpaired, 2-tailed Student t test. ***P < .001. (D) MA plot showing upregulation and downregulation of genes in native Mpig6bmut MKs derived from female mice compared with female WT control mice. Black lines point toward downregulated MK-specific genes (eg, Tubb1, Gp6, and Gp1ba). (E) Upregulation and downregulation of MK-associated genes and non-megakaryocytic blood lineage markers in native MKs from female Mpig6bmut mice compared with the respective control (n = 4). Only values with a log2-fold change <1.0 (dotted line) were considered upregulated or downregulated. Cd3d, CD3 δ chain; Csk, C-Src kinase; Fli1, friend leukemia integration 1 transcription factor; Gata1, GATA-binding factor 1; Gfi1b, growth factor-independent 1B transcriptional repressor; Hba-a1, hemoglobin subunit α; Ikzf5, Ikaros family zinc finger protein 5; Matk, megakaryocyte-associated tyrosine-protein kinase; Mpl, myeloproliferative leukemia protein; Mpo, myeloperoxidase; Runx1, runt-related transcriptions factor 1; Stat3, signal transducer and activator of transcription 3. (F) Immunostainings of femora cryosections visualizing GATA-1 expression in WT and Mpig6bmut MKs in situ. Images are representative of 10 fields of view (FOV) per mouse (n = 6). Bars represent 50 µm. (G) Quantification of phosphorylation and/or total levels of GATA-1, Jak2, STAT5a/b, and Shp2 in in vitro–differentiated starved or TPO-stimulated WT and Mpig6bmut MKs analyzed using an automated quantitative capillary-based immunoassay platform; Jess (ProteinSimple). Corresponding representative blots are shown in supplemental Figure 3F. Values are mean ± SD (n = 5). One-way analysis of variance with Sidak correction for multiple comparisons. *P < .05; **P < .01; ***P < .001. (H) Mean size distribution of WT and Mpig6b−/− MKs was analyzed by flow cytometry. Values are mean ± SD (n = 2). Unpaired, 2-tailed Student t test. *P < .05.
Maturation block in Mpig6bmut MKs involves reduced gene expression and TPO signaling. (A-B) Mean size distribution of WT and Mpig6bmut MKs was analyzed by flow cytometry. (A) Dot plots depicting the proportion of SSChigh MKs and the delineation between small, medium, and large MKs. (B) Values are mean ± SD (n = 6). Unpaired, 2-tailed Student t test. *P < .05; **P < .01; ***P < .001. (C) Mean surface receptor expression on the whole MK population derived from WT and Mpig6bmut mice. Values are mean ± SD (n = 4). Unpaired, 2-tailed Student t test. ***P < .001. (D) MA plot showing upregulation and downregulation of genes in native Mpig6bmut MKs derived from female mice compared with female WT control mice. Black lines point toward downregulated MK-specific genes (eg, Tubb1, Gp6, and Gp1ba). (E) Upregulation and downregulation of MK-associated genes and non-megakaryocytic blood lineage markers in native MKs from female Mpig6bmut mice compared with the respective control (n = 4). Only values with a log2-fold change <1.0 (dotted line) were considered upregulated or downregulated. Cd3d, CD3 δ chain; Csk, C-Src kinase; Fli1, friend leukemia integration 1 transcription factor; Gata1, GATA-binding factor 1; Gfi1b, growth factor-independent 1B transcriptional repressor; Hba-a1, hemoglobin subunit α; Ikzf5, Ikaros family zinc finger protein 5; Matk, megakaryocyte-associated tyrosine-protein kinase; Mpl, myeloproliferative leukemia protein; Mpo, myeloperoxidase; Runx1, runt-related transcriptions factor 1; Stat3, signal transducer and activator of transcription 3. (F) Immunostainings of femora cryosections visualizing GATA-1 expression in WT and Mpig6bmut MKs in situ. Images are representative of 10 fields of view (FOV) per mouse (n = 6). Bars represent 50 µm. (G) Quantification of phosphorylation and/or total levels of GATA-1, Jak2, STAT5a/b, and Shp2 in in vitro–differentiated starved or TPO-stimulated WT and Mpig6bmut MKs analyzed using an automated quantitative capillary-based immunoassay platform; Jess (ProteinSimple). Corresponding representative blots are shown in supplemental Figure 3F. Values are mean ± SD (n = 5). One-way analysis of variance with Sidak correction for multiple comparisons. *P < .05; **P < .01; ***P < .001. (H) Mean size distribution of WT and Mpig6b−/− MKs was analyzed by flow cytometry. Values are mean ± SD (n = 2). Unpaired, 2-tailed Student t test. *P < .05.
Defective MK-specific gene expression in Mpig6bmut mice
Next, we performed bulk RNA sequencing on a population of purified BM-derived native MKs from young adult mutant mice, not yet displaying any signs of myelofibrosis in comparison with WT littermate controls. Our protocol used an anti-CD61 antibody coupled to magnetic microbeads followed by a bovine serum albumin–density gradient, similar to the isolation protocol by Davizon-Castillo et al.18 The transcriptome signature of Mpig6bmut MKs revealed a striking decrease in a plethora of MK-specific transcripts, including Tubb1, Myl9, and Gp1ba (Figure 2D). The data confirmed that the single-nucleotide exchange within Mpig6b results in the lack of Mpig6b mRNA expression. mRNA levels of transcription factors GATA1 and Gfi1b, which are both indispensable for MK differentiation,19,20 were significantly less abundantly expressed in Mpig6bmut MKs (Figure 2E; supplemental Figure 3D). Notably, the expression levels of several MK-associated transcripts (Csk, Matk,21 and Syk) including transcription factors (Runx1, Fli1, Stat3, Stat5a/b, and Ikzf5) were overall unaltered, confirming similar purification of WT and mutant cells. The total transcript numbers of non-megakaryocytic blood lineage markers (including Cd3d, Cd19, Mpo, and Hba-a1) was very low and comparable between WT and mutant MKs, independently corroborating the high purity in our MK isolation approach (Figure 2E; supplemental Figure 3D). Quantitative polymerase chain reaction analysis revealed that transcript levels of Itgb3, Tubb1, and Gata1 were also significantly reduced when mutant MKs were in vitro–differentiated (supplemental Figure 3E). In addition, GATA-1 protein levels were reduced in both native MKs in situ (Figure 2F), as well as in in vitro–differentiated Mpig6bmut MKs (Figure 2G; supplemental Figure 3F). Our RNA-sequencing data strongly implies that loss of G6b-B hampers early transcriptional pathways within the megakaryocytic lineage, as reflected by downregulation of many MK-specific genes in primary MKs.
Defective TPO-signaling in Mpig6bmut MKs
We thus investigated the total MK protein and phosphorylation levels of proteins that are crucial to TPO signaling,11,22 Protein levels of c-Mpl and Jak2 decreased in Mpig6bmut MKs compared with WT MKs, whereas STAT5a/b were not significantly altered. Moreover, c-Mpl, Jak2, and STAT5a/b phosphorylation was markedly reduced (Figure 2G; supplemental Figure 3F). Although the underlying molecular mechanism requires further investigation, it may involve the timely recruitment of the protein tyrosine phosphatases Shp1 and/or Shp2 by G6b-B2,23,24 to a specific substrate protein(s) downstream of the c-Mpl receptor. Shp2 is known to play an important role in this pathway,11,22 and we found reduced protein levels and phosphorylation of Shp2 (Figure 2G; supplemental Figure 3F). Our findings thus provide evidence that loss of G6b-B disturbs TPO-based signaling events in MKs and point to a critical role of G6b-B in regulating relevant signaling pathways driving MK maturation.
Mpig6b−/− mice display less mature MKs
To corroborate that our findings reflect the role of G6b-B and are not the consequence of an unlikely cosegregating mutation, we generated a novel Mpig6b−/− mouse model (supplemental Figure 4A-D). MKs from this novel Mpig6b−/− mice displayed an increase in the percentage of small- and medium-sized MKs and a marked decrease in large and SSChigh MKs (Figure 2H), providing an additional layer of evidence that the lack of G6b-B leads to an MK maturation defect.
Patients with disease-causing variants within MPIG6B present with congenital macrothrombocytopenia, mild-to-moderate bleeding diathesis, focal myelofibrosis, and atypical MKs; however, the underlying causes of the disease have remained unclear.3-7 Our results demonstrate a previously unrecognized key function of G6b-B in MK maturation by regulating cell size, demarcation membrane system development, and gene expression. These findings put previous results into new context, suggesting that the reduced surface expression of receptors on the whole MK population is not a consequence of increased shedding,2,11 but the reduced proportion of mature cells. We propose that the severe macrothrombocytopenia in Mpig6bmut mice is a direct consequence of the unexpected overall MK maturation defect. We observed an increased number of immature MKs in the bone marrow, which may help to better understand the cause of myelofibrosis in G6b-B–null mice and patients, as an accumulation of immature MKs has been described as the main driver of this complex disease.25-29
Acknowledgments
The authors thank Stefanie Hartmann, Sylvia Hengst, Daniela Naumann, and Mariola Dragan for excellent technical assistance; the microscopy platform of the Bioimaging Center Würzburg for providing technical infrastructure and support; Yotis Senis (Strasbourg, France) for providing anti-G6b-B antibodies; and the Core Unit SysMed at the University of Würzburg for excellent technical support and RNA-seq data generation.
This work was supported by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) Project 374031971-TRR 240/Project A01 (B.N.), Project A03 (H.S.), and grant NI 556/11-2 (B.N.); the Interdisciplinary Center for Clinical Research (IZKF) at the University of Würzburg (project Z-6) the European Union (EFRE; Europäischer Fonds für Regionale Entwicklung, Bavaria, Germany), and a German Excellence Initiative grant to the Graduate School of Life Sciences, University of Würzburg (I.C.B. and Z.N.); and the MINT program of the Hanns-Seidel-Stiftung (S.S.).
Authorship
Contribution: I.C.B., Z.N., G.M., H.S., and B.N. conceived the study; M.H.-L., R.B., and S.D. developed the methodology; T.H., R.B., and S.D. created the software; I.C.B., Z.N., G.M., M.H.-L., M.E., T.H., T.V., C.G., K.M., J.H., S.S., and A.B. conducted the investigation; T.S., N.S., T.H., D.S., H.S., and B.N. provided the resources; I.P., A.I., H.S., and B.N. supervised the study; I.C.B., Z.N., G.M., H.S., and B.N. wrote the original manuscript; and I.C.B., Z.N., G.M., M.H.-L., I.P., H.S., and B.N. wrote, reviewed, and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing interests.
The current affiliation for I.C.B. is Vascular Biology Program, Boston Children’s Hospital and Harvard Medical School, Boston, MA.
Correspondence: Bernhard Nieswandt, University Hospital, University of Würzburg, Josef-Schneider-Straße 2, 97080 Würzburg, Germany; e-mail: bernhard.nieswandt@virchow.uni-wuerzburg.de; or Harald Schulze, University Hospital, University of Würzburg, Josef-Schneider-Straße 2, 97080 Würzburg, Germany; e-mail: harald.schulze@uni-wuerzburg.de.

