

Key Points
A 20-residue region in domain 6 of HK plays a critical role in Aβ42-induced contact system activation.
3E8 blocks PK and FXI binding to HK, disassembles HK/PK and HK/FXI complexes, and delays intrinsic coagulation.

Abstract
Alzheimer’s disease (AD) is a neurodegenerative disorder and the leading cause of dementia. Vascular abnormalities and neuroinflammation play roles in AD pathogenesis. Plasma contact activation, which leads to fibrin clot formation and bradykinin release, is elevated in many AD patients, likely due to the ability of AD’s pathogenic peptide β-amyloid (Aβ) to induce its activation. Since overactivation of this system may be deleterious to AD patients, the development of inhibitors could be beneficial. Here, we show that 3E8, an antibody against a 20-amino acid region in domain 6 of high molecular weight kininogen (HK), inhibits Aβ-induced intrinsic coagulation. Mechanistically, 3E8 inhibits contact system activation by blocking the binding of prekallikrein (PK) and factor XI (FXI) to HK, thereby preventing their activation and the continued activation of factor XII (FXII). The 3E8 antibody can also disassemble HK/PK and HK/FXI complexes in normal human plasma in the absence of a contact system activator due to its strong binding affinity for HK, indicating its prophylactic ability. Furthermore, the binding of Aβ to both FXII and HK is critical for Aβ-mediated contact system activation. These results suggest that a 20-amino acid region in domain 6 of HK plays a critical role in Aβ-induced contact system activation, and this region may provide an effective strategy to inhibit or prevent contact system activation in related disorders.
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder and a major cause of dementia. The pathological hallmarks of AD are β-amyloid (Aβ) plaques, tau tangles, neuroinflammation, and neuronal degeneration. There is evidence that vascular dysfunction is also a core pathology of AD.1-3 Not only is disruption of the blood-brain barrier (BBB) an early biomarker of cognitive dysfunction,4 but cerebrovascular and blood flow abnormalities are also associated with cognitive decline and AD progression.5-7 AD is a complex and heterogeneous disease, and it is unlikely that only one mechanism can be targeted for treatment of all patients.8 It is therefore important to identify various pathogenic pathways which will allow stratification of patients and personalized treatment.
Dysfunctional blood clotting is often observed in AD patients and mouse models.7,9-18 Parenchymal brain deposits of fibrin, a proinflammatory protein that is also the main component of blood clots,19,20 play a significant role in AD progression in mouse models.9,10,19,21-24 Fibrin and its precursor, fibrinogen, are blood proteins, and their presence in the brain indicates a loss of BBB integrity in these patients. Fibrin is formed upon the generation of thrombin during the clotting pathway of the contact activation system.
The plasma contact system, which includes both clotting and inflammatory pathways,25 may play a role in AD pathogenesis.14,26 Aβ42, one of the primary pathogenic factors of AD,27-29 can activate factor XII (FXII) to initiate the contact system.30-33 Activated FXII (FXIIa) can activate factor XI (FXI) to trigger the intrinsic clotting pathway, leading to thrombin generation and fibrin formation,32,34 whereas FXIIa activation of prekallikrein (PK) leads to the release of bradykinin from its precursor high molecular weight kininogen (HK) and subsequent activation of inflammatory processes.6,32,35-39 Both fibrin formation and bradykinin-mediated inflammation may contribute to AD.9,18,22 It has been shown that the AD brain parenchyma exhibits higher plasma kallikrein (PKa) activity,40 and Aβ plaques contain FXII41 and bradykinin.18 Compared with nondemented individuals, AD patients have increased levels of cleaved HK (cHK) in their cerebrospinal fluid42 and higher plasma levels of FXIIa, cHK, and bradykinin.15,18,43 In a mouse model of AD, plasma contact system activation is also elevated,43 and knockdown of this system using an anti-FXII antisense oligonucleotide ameliorates AD pathology and cognitive impairment in a mouse model.35 Therefore, inhibition of the contact system could be beneficial for AD patients.
We previously showed that an anti-HK antibody (clone 3E8, raised against a 20-amino acid region in domain 6 of HK) blocks Aβ42-induced HK cleavage and bradykinin release.36 Our new studies revealed that 3E8 also prevents Aβ42-induced intrinsic coagulation. Mechanistically, 3E8 inhibits contact system activation by blocking the binding of PK and FXI to HK, thereby preventing activation of PK and FXI and continued activation of FXII. We also show here that 3E8 disassembles HK/PK and HK/FXI complexes in normal human plasma in the absence of a contact system activator, suggesting its prophylactic ability. Moreover, our results revealed that Aβ42 can bind HK, which is critical for Aβ42-mediated contact system activation. Therefore, our results show for the first time that a 20-amino acid region of HK plays an essential role in Aβ42-mediated contact system activation, and targeting this region may provide an effective strategy to treat contact system-related pathological conditions such as AD.
Materials and methods
Preparation of anti-HK antibodies and Aβ42
Binding experiments
The binding experiments were performed using enzyme-linked immunosorbent assay (ELISA).45 For 3E8 binding to HK, plates were coated with 2 μg/mL 3E8 in binding buffer (0.1 M sodium bicarbonate; pH 9.6). For PK and FXI binding to HK, plates were coated with 2 μg/mL of PK or FXI in binding buffer, respectively. The plates were then incubated with various concentrations of HK in the binding buffer. Bound HK was detected with an antibody to HK as described.44 The dissociation constant (KD) of HK and 3E8 interaction was determined from the binding curve using a nonlinear regression curve fit equation in GraphPad Prism software. For 3E8 blocking, PK/HK binding, or FXI/HK binding studies, HK was preincubated at 37°C in the absence or presence of increasing concentrations of the 3E8 antibody. Bound HK was then determined as described above.
Blood collection and plasma preparation
The collection and preparation of human plasma were approved by The Rockefeller University Institutional Review Board. Blood from healthy human donors (n = 6) who had given informed, written consent was collected at The Rockefeller University Hospital. Plasma was prepared as described previously.44 Pooled normal human plasma (NHP) and FXII-deficient (FXII-DF) human plasma were purchased from George King Bio-Medical. Kininogen-deficient (KN-DF) human plasma was purchased from Technoclone.
Aβ42 pulldown experiments and anti-HK antibody immunoprecipitation
NHP, FXII-DF, or KN-DF human plasma was incubated with biotinylated Aβ42 (B-Aβ42) in the presence or absence of 3E8 anti-HK antibody. Dynabeads M-280 Streptavidin (Invitrogen) was used to pull down the B-Aβ42/bound proteins complex according to the manufacturer’s instructions.
For anti-HK antibody immunoprecipitation, anti-HK antibodies (3E8, 2B7, 4B12)44 and control immunoglobulin G (IgG) (Innovative Research) were biotinylated using EZ-Link Sulfo-NHS-LC-Biotin (Thermo Scientific) according to the manufacturer’s instructions. Ethylenediaminetetraacetic acid (EDTA) plasma was incubated with biotinylated HK antibodies and control IgG. In 2B7 anti-HK antibody pulldown experiments, NHP was incubated with 3E8/phosphate-buffered saline (PBS)/IgG, then incubated with B-2B7 at 37°C for 20 minutes. Dynabeads M-280 Streptavidin was used to pull down the antibody-antigen complex. Samples were eluted with sodium dodecyl sulfate (SDS) sample buffer, and western blots were performed.
HK D6 peptide competition experiments
A 20-residue peptide derived from domain 6 of HK (IQSDDDWIPDIQIDPNGLSF) was synthesized at Rockefeller’s Proteomics Resource Center. This peptide (hereafter referred to as HK-D6) was used as an antigen to generate the 3E8 anti-HK antibody. Human plasma was incubated with buffer; 3E8 anti-HK antibody (0.5 μM); control IgG (0.5 μM); HK-D6 (0.5 μM, 1 μM, 2μM); or 3E8 anti-HK antibody or control IgG (0.5 μM) + HK-D6 (0.5 μM, 1 μM, 2 μM). Incubations were left at 37°C for 20 minutes, and then Aβ42 (5 μM) was added and incubated for 2 hours. Western blotting analyses were performed.
Western blotting
Western blots were performed as described previously.46 Plasmas after various treatments were heated to 95°C for 5 minutes in sample buffer. Equal amounts of plasma from each sample were run on SDS polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene difluoride membrane (Bio-Rad), and analyzed by western blot with antibodies against FXII/FXIIa (Cedarlane Laboratories), FXI (Haematologic Technologies), PK (Affinity Biologicals), HK and cHK (3E8), HK light chain (Abcam), low molecular weight kininogen (LMWK) (Novus Biologicals), and transferrin (TF) (Abcam). Blots were imaged via Bio-Rad ChemiDoc. Protein levels were quantified by densitometry with the National Institutes of Health Image J. All experiments used plasmas from 6 healthy donors, were run in duplicate, and repeated at least 3 times.
Plasma kallikrein activity
EDTA-plasma was incubated with buffer, control IgG (50 nM), or various concentrations of 3E8 anti-HK antibody (5 nM, 25 nM, and 50 nM) at 37°C for 20 minutes in plates. Aβ42 (5 μM) and chromogenic substrate S-2302 (0.67 mM final concentration, Diapharma) were added, and absorbance at 405 nm was read for 60 minutes at 37°C in a spectrophotometer (Molecular Devices). Plasmas from 6 different human donors were used, and each plasma was used twice.
Activated partial thromboplastin time (aPTT) and prothrombin time (PT) tests in human plasma
A spectrophotometer-based aPTT assay was performed as described previously15 with some modifications. Briefly, citrated plasma was warmed at 37°C for 3 minutes and incubated with buffer or control IgG (3.75 μM) or various concentrations of 3E8 anti-HK antibody (0.05 μM, 0.15 μM, 0.75 μM, and 3.75 μM) at 37°C for 20 minutes in a 96-well plate. Then aPTT reagent solution (30 μl; APTT-XL; Pacific Hemostasis) was added and incubated for 5 minutes. Clot formation was initiated by adding 30 μl of 25 mM CaCl2 solution. Clot formation was monitored kinetically at 350 nm at 37°C over time using SoftMax Pro 6.1 software (Molecular Devices) as described previously.15
For the effects of 3E8 anti-HK antibody on Aβ42-induced intrinsic coagulation changes, citrated plasma was incubated with buffer or control IgG (3.75 μM) or various concentrations of 3E8 (0.05 μM, 0.15 μM, 0.75 μM, and 3.75 μM) at 37°C for 20 minutes. Aβ42 (4 μM) was added and incubated at 37°C for 10 minutes, then CaCl2 solution was added, and clotting was determined as described above.
For PT, citrated plasma was warmed to 37°C for 3 minutes and incubated with buffer, control IgG, or 3E8 anti-HK antibody (3.75 μM) at 37°C for 20 minutes in 96-well plates. Clotting was monitored kinetically as described above after the addition of PT reagent Thromboplastin D (Pacific Hemostasis).
Statistical analyses
All statistical analyses were performed using GraphPad Prism 4 software. Comparisons among multiple groups were performed using 1-way ANOVA followed by Newman-Keuls multiple comparison test.
Results
Characterization of the 3E8 anti-HK antibody
The 3E8 anti-HK antibody dose-dependently blocks Aβ42-induced HK cleavage and bradykinin release in human plasma ex vivo.36 To investigate the specificity of this antibody, we performed western blotting with purified proteins and human plasmas and probed with 3E8 (Figure 1A). 3E8 detected purified human HK and cHK, and it showed a single band corresponding to HK in NHP and a single band corresponding to cHK in Aβ42-treated human plasma. There were no bands detected in KN-DF plasma. When the membrane was stripped and reprobed with an antibody against LMWK, LMWK was detected in NHP and Aβ42-treated human plasma, but not in KN-DF plasma (Figure 1A). This LMWK band was not revealed by the 3E8 anti-HK antibody, indicating 3E8 does not recognize LMWK and is specific for HK and cHK.
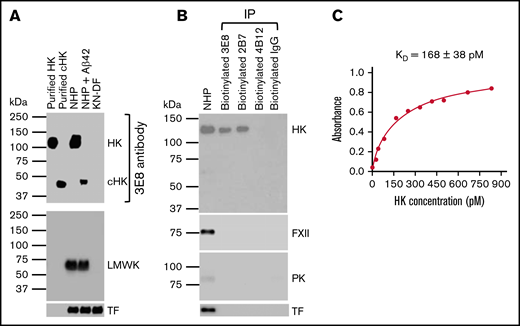
Characterization of the 3E8 anti-HK antibody. (A) Western blot shows 3E8 specifically recognizes HK in NHP and cHK in Aβ42-treated human plasma. Purified human HK and cHK were included as controls. In KN-DF human plasma, HK was not detected by 3E8, indicating the specificity of 3E8. The membrane was stripped and reprobed with anti-LMWK antibody. LMWK was detected in normal and Aβ42-treated human plasma but not in KN-DF plasma. 3E8 did not recognize LMWK. (B) The 3E8 and 2B7 anti-HK antibodies pulled down HK, but not FXII, PK, or TF from human plasma. Human plasma was incubated with biotinylated 3E8, 2B7, and 4B12 anti-HK antibodies and control IgG, and streptavidin was added to pull down the antibody-antigen complex. The samples were analyzed by western blot using commercial antibodies against HK, FXII, PK, and TF. The 3E8 and 2B7 HK antibodies immunoprecipitated HK from human plasma, but they did not pull down FXII, PK, or TF. The 4B12 antibody, which recognizes cleaved HK but not intact HK, did not pull down HK, FXII, PK, or TF from human plasma. (C) HK ELISA shows the KD of 3E8 anti-HK antibody binding to HK is 168 ± 38 pM. The experiments were performed in triplicate and repeated 3 times.
Characterization of the 3E8 anti-HK antibody. (A) Western blot shows 3E8 specifically recognizes HK in NHP and cHK in Aβ42-treated human plasma. Purified human HK and cHK were included as controls. In KN-DF human plasma, HK was not detected by 3E8, indicating the specificity of 3E8. The membrane was stripped and reprobed with anti-LMWK antibody. LMWK was detected in normal and Aβ42-treated human plasma but not in KN-DF plasma. 3E8 did not recognize LMWK. (B) The 3E8 and 2B7 anti-HK antibodies pulled down HK, but not FXII, PK, or TF from human plasma. Human plasma was incubated with biotinylated 3E8, 2B7, and 4B12 anti-HK antibodies and control IgG, and streptavidin was added to pull down the antibody-antigen complex. The samples were analyzed by western blot using commercial antibodies against HK, FXII, PK, and TF. The 3E8 and 2B7 HK antibodies immunoprecipitated HK from human plasma, but they did not pull down FXII, PK, or TF. The 4B12 antibody, which recognizes cleaved HK but not intact HK, did not pull down HK, FXII, PK, or TF from human plasma. (C) HK ELISA shows the KD of 3E8 anti-HK antibody binding to HK is 168 ± 38 pM. The experiments were performed in triplicate and repeated 3 times.
We also performed immunoprecipitation using 3E8, 2B7 (intact HK-specific), and 4B12 (cHK-specific) antibodies.44 Both 3E8 and 2B7 immunoprecipitated intact HK, but not FXII or PK, from NHP (Figure 1B), indicating that 3E8 and 2B7 do not bind FXII or PK. Neither 4B12 nor control IgG pulled down intact HK from NHP. This experiment showed that 3E8 binds HK but not other contact system proteins in human plasma. Furthermore, using ELISA,45 we determined that the KD of 3E8 for HK is 168 ± 38 pM, which indicates a very strong binding affinity (Figure 1C).
The inhibitory effects of 3E8 anti-HK antibody on Aβ42-induced HK cleavage are abolished by the competitive HK-D6 peptide
The 3E8 anti-HK antibody was generated using a peptide derived from D6 of human HK. To analyze whether 3E8 blocks HK cleavage by binding specifically to the D6 of HK in human plasma, we examined the inhibitory effects of 3E8 on HK cleavage in the presence of various concentrations of HK-D6 (Figure 2). Aβ42 induced HK cleavage, and HK cleavage was blocked by 3E8 (lanes 2 and 3, Figure 2A). Under the same conditions, HK-D6 did not show much effect on Aβ42-induced HK cleavage (lanes 5-7, Figure 2A). However, HK-D6 dose-dependently blocked the inhibitory effects of 3E8 on Aβ42-induced HK cleavage (lanes 8-10, Figure 2A). Control IgG and IgG+HK-D6 did not show any effect on Aβ42-induced HK cleavage (lanes 4 and 11-13, Figure 2A). These results suggest that the inhibitory effect on HK cleavage by 3E8 is dose-dependently abolished by the competitive HK-D6 peptide (Figure 2B), confirming that 3E8 blocks HK cleavage by binding to this 20-amino acid region of D6 in HK (antigen of 3E8) in human plasma.
![The inhibitory effects of the 3E8 anti-HK antibody on Aβ42-induced HK cleavage are abolished by competitive HK-D6 peptides. (A) EDTA-human plasma was incubated with buffer, 3E8 anti-HK antibody (0.5 μM), control IgG (0.5 μM), HK-D6 (0.5 μM, 1 μM, 2 μM), 3E8 anti-HK antibody (0.5 μM) + HK-D6 (0.5 μM, 1 μM, 2 μM) or control IgG (0.5 μM) + HK-D6 (0.5 μM, 1 μM, 2 μM) (as indicated in [A]) at 37°C for 20 minutes. Aβ42 (5 μM) was added and incubated for 2 hours. Western blotting analyses were performed with antibodies against HK and TF. 3E8 HK antibody protected HK from Aβ42-induced cleavage (lane 3), but control IgG (lane 4) and different concentrations of HK-D6 did not have significant effects on Aβ42-induced HK cleavage (lanes 5-7 and [B]). However, HK-D6 dose-dependently inhibited the protective effects of 3E8 anti-HK antibody on Aβ42-induced HK cleavage (lanes 1, 3, 8-10, and [B]). HK-D6 did not have significant effects on the control IgG in Aβ42-induced HK cleavage (lanes 4, 11-13, and [B]). HK western blotting signals were normalized to TF. n = 6. Data are denoted as mean ± SEM. *P < .05, **P ≤ .01, ***P ≤ .001. P > .05 was not significant (n.s.).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/10/10.1182_bloodadvances.2021006612/3/m_advancesadv2021006612f2.png?Expires=1765894666&Signature=zPbhxvJXpJ3rWvMYfP0bY3kmbz18dcqTBg1tUkuM8UUkYY-f5QZwH~2Y4AT96DA7iX5pJyqN2ADGZgodQCnyE-aA~VJZ~GH~JvDx80QLcYwWS8ILLgmY-OfxT70eYW3qq0ebRPRgonxNbcnG7gFrpWllPpEBac-8lOvJCjUeAcGAKN9AiPvZTJmeXHHJXEXc8fWp6hieIc1PuSVlrzNLtn6JRvim-6ldhMMSF36ap7m54EGDXGw~-8KXsyJVBg3XTY3NdUUr9sYakdPzCTdOZcoN7FTuL-uCVTpmj4-339NNvPqOXE6Ni44KLSsO501ZbFV970r9PWkLzY~HmdIY4g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The inhibitory effects of the 3E8 anti-HK antibody on Aβ42-induced HK cleavage are abolished by competitive HK-D6 peptides. (A) EDTA-human plasma was incubated with buffer, 3E8 anti-HK antibody (0.5 μM), control IgG (0.5 μM), HK-D6 (0.5 μM, 1 μM, 2 μM), 3E8 anti-HK antibody (0.5 μM) + HK-D6 (0.5 μM, 1 μM, 2 μM) or control IgG (0.5 μM) + HK-D6 (0.5 μM, 1 μM, 2 μM) (as indicated in [A]) at 37°C for 20 minutes. Aβ42 (5 μM) was added and incubated for 2 hours. Western blotting analyses were performed with antibodies against HK and TF. 3E8 HK antibody protected HK from Aβ42-induced cleavage (lane 3), but control IgG (lane 4) and different concentrations of HK-D6 did not have significant effects on Aβ42-induced HK cleavage (lanes 5-7 and [B]). However, HK-D6 dose-dependently inhibited the protective effects of 3E8 anti-HK antibody on Aβ42-induced HK cleavage (lanes 1, 3, 8-10, and [B]). HK-D6 did not have significant effects on the control IgG in Aβ42-induced HK cleavage (lanes 4, 11-13, and [B]). HK western blotting signals were normalized to TF. n = 6. Data are denoted as mean ± SEM. *P < .05, **P ≤ .01, ***P ≤ .001. P > .05 was not significant (n.s.).
The inhibitory effects of the 3E8 anti-HK antibody on Aβ42-induced HK cleavage are abolished by competitive HK-D6 peptides. (A) EDTA-human plasma was incubated with buffer, 3E8 anti-HK antibody (0.5 μM), control IgG (0.5 μM), HK-D6 (0.5 μM, 1 μM, 2 μM), 3E8 anti-HK antibody (0.5 μM) + HK-D6 (0.5 μM, 1 μM, 2 μM) or control IgG (0.5 μM) + HK-D6 (0.5 μM, 1 μM, 2 μM) (as indicated in [A]) at 37°C for 20 minutes. Aβ42 (5 μM) was added and incubated for 2 hours. Western blotting analyses were performed with antibodies against HK and TF. 3E8 HK antibody protected HK from Aβ42-induced cleavage (lane 3), but control IgG (lane 4) and different concentrations of HK-D6 did not have significant effects on Aβ42-induced HK cleavage (lanes 5-7 and [B]). However, HK-D6 dose-dependently inhibited the protective effects of 3E8 anti-HK antibody on Aβ42-induced HK cleavage (lanes 1, 3, 8-10, and [B]). HK-D6 did not have significant effects on the control IgG in Aβ42-induced HK cleavage (lanes 4, 11-13, and [B]). HK western blotting signals were normalized to TF. n = 6. Data are denoted as mean ± SEM. *P < .05, **P ≤ .01, ***P ≤ .001. P > .05 was not significant (n.s.).
3E8 dose-dependently inhibits Aβ42-induced PK and FXII activation
The 3E8 anti-HK antibody binds to the D6 of HK, the region that binds PK and FXI, and hence plays an important role in contact system activation.47-50 We analyzed whether 3E8 impacts PK activation. 3E8 dose-dependently inhibited Aβ42-induced HK cleavage36 as well as PK activation (Figure 3A-B). We also analyzed whether 3E8 inhibits Aβ42-induced kallikrein activity using a chromogenic substrate. In the absence of 3E8, Aβ42 induced kallikrein generation in NHP. However, 3E8 dose-dependently inhibited Aβ42-induced kallikrein activation (Figure 3C). Control IgG had no effect on kallikrein activity.
![3E8 dose-dependently inhibits Aβ42-induced PK and FXII activation in human plasma. (A,B) The 3E8 anti-HK antibody dose-dependently inhibited Aβ42-induced PK activation in NHP. NHP was incubated with buffer, 3E8 anti-HK antibody (0.02 μM, 0.1 μM, 0.5 μM), or control IgG (0.5 μM) at 37°C for 20 minutes. Aβ42 (5 μM) was added and incubated at 37°C for 2 hours (A). Western blots were performed with antibodies against HK, PK, FXII, and TF. 3E8 dose-dependently protected HK from Aβ42-induced cleavage, as shown previously.36 It also dose-dependently inhibited PK activation. Decreases in the PK band indicate activation of PK to PKa; PKa was not detectable by this antibody (lanes 1-5 in [A-B]). Control IgG did not show any effect on Aβ42-induced PK activation (lane 6 in [A-B]). (C) 3E8 dose-dependently inhibited Aβ42-induced kallikrein activity in human plasma. NHP was incubated with buffer, control IgG (50 nM), or various concentrations of 3E8 HK antibody (5 nM, 25 nM, and 50 nM) at 37°C for 20 minutes. Aβ42 (5 μM) and chromogenic substrate S-2302 (0.67 mM final concentration) were added, and absorbance at 405 nm was read for 60 minutes at 37°C. In the absence of 3E8, Aβ42 induced a dramatic increase in kallikrein activity. The 3E8 antibody dose-dependently inhibited Aβ42-induced kallikrein activation, while control IgG had no effect. (D-E) 3E8 anti-HK antibody dose-dependently inhibited FXII activation. 3E8 also dose-dependently inhibited FXII activation (FXIIa) (lanes 1-5 in [D-E]), while control IgG did not influence Aβ42-induced FXII activation (lane 6 in [D-E]). FXIIa and PK western blots were normalized against TF. n = 6. Data are denoted as mean ± SEM. *P < .05, **P ≤ .01, ***P ≤ .001. P > .05 was not significant (n.s.).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/10/10.1182_bloodadvances.2021006612/3/m_advancesadv2021006612f3.png?Expires=1765894666&Signature=SLqcWGMWyt4-X9D11Ckych4QdUydGqE6-G1LTFphYI3B0BJgRIGeCwoWpvnHslMHAKKM7SVwmv1Xp19HEdqRnkkx0z9Gc-EOKUst9mnEQNdn0OxkUq1rjz0LxY~FCMNYzYt8oop5HfaxSiG0dWYqDr-HhbaHXQDMV5uCEZsKSzmbLyCXIbPEvhjck4WGV3CQ6th0dAaJIi28Aa3YKtslB6DhowCDGREb9z3yHXOJfkDs5r-lA09slqDSHy6E5JIWQbhD1JL5B8cyOw4kO~lZXmzY5Hq7u7yJRtcCEjLisnF-yAtNSU9i3TMjPQmuuYFJZeH3RSd3ORDI11N7bTsOPA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
3E8 dose-dependently inhibits Aβ42-induced PK and FXII activation in human plasma. (A,B) The 3E8 anti-HK antibody dose-dependently inhibited Aβ42-induced PK activation in NHP. NHP was incubated with buffer, 3E8 anti-HK antibody (0.02 μM, 0.1 μM, 0.5 μM), or control IgG (0.5 μM) at 37°C for 20 minutes. Aβ42 (5 μM) was added and incubated at 37°C for 2 hours (A). Western blots were performed with antibodies against HK, PK, FXII, and TF. 3E8 dose-dependently protected HK from Aβ42-induced cleavage, as shown previously.36 It also dose-dependently inhibited PK activation. Decreases in the PK band indicate activation of PK to PKa; PKa was not detectable by this antibody (lanes 1-5 in [A-B]). Control IgG did not show any effect on Aβ42-induced PK activation (lane 6 in [A-B]). (C) 3E8 dose-dependently inhibited Aβ42-induced kallikrein activity in human plasma. NHP was incubated with buffer, control IgG (50 nM), or various concentrations of 3E8 HK antibody (5 nM, 25 nM, and 50 nM) at 37°C for 20 minutes. Aβ42 (5 μM) and chromogenic substrate S-2302 (0.67 mM final concentration) were added, and absorbance at 405 nm was read for 60 minutes at 37°C. In the absence of 3E8, Aβ42 induced a dramatic increase in kallikrein activity. The 3E8 antibody dose-dependently inhibited Aβ42-induced kallikrein activation, while control IgG had no effect. (D-E) 3E8 anti-HK antibody dose-dependently inhibited FXII activation. 3E8 also dose-dependently inhibited FXII activation (FXIIa) (lanes 1-5 in [D-E]), while control IgG did not influence Aβ42-induced FXII activation (lane 6 in [D-E]). FXIIa and PK western blots were normalized against TF. n = 6. Data are denoted as mean ± SEM. *P < .05, **P ≤ .01, ***P ≤ .001. P > .05 was not significant (n.s.).
3E8 dose-dependently inhibits Aβ42-induced PK and FXII activation in human plasma. (A,B) The 3E8 anti-HK antibody dose-dependently inhibited Aβ42-induced PK activation in NHP. NHP was incubated with buffer, 3E8 anti-HK antibody (0.02 μM, 0.1 μM, 0.5 μM), or control IgG (0.5 μM) at 37°C for 20 minutes. Aβ42 (5 μM) was added and incubated at 37°C for 2 hours (A). Western blots were performed with antibodies against HK, PK, FXII, and TF. 3E8 dose-dependently protected HK from Aβ42-induced cleavage, as shown previously.36 It also dose-dependently inhibited PK activation. Decreases in the PK band indicate activation of PK to PKa; PKa was not detectable by this antibody (lanes 1-5 in [A-B]). Control IgG did not show any effect on Aβ42-induced PK activation (lane 6 in [A-B]). (C) 3E8 dose-dependently inhibited Aβ42-induced kallikrein activity in human plasma. NHP was incubated with buffer, control IgG (50 nM), or various concentrations of 3E8 HK antibody (5 nM, 25 nM, and 50 nM) at 37°C for 20 minutes. Aβ42 (5 μM) and chromogenic substrate S-2302 (0.67 mM final concentration) were added, and absorbance at 405 nm was read for 60 minutes at 37°C. In the absence of 3E8, Aβ42 induced a dramatic increase in kallikrein activity. The 3E8 antibody dose-dependently inhibited Aβ42-induced kallikrein activation, while control IgG had no effect. (D-E) 3E8 anti-HK antibody dose-dependently inhibited FXII activation. 3E8 also dose-dependently inhibited FXII activation (FXIIa) (lanes 1-5 in [D-E]), while control IgG did not influence Aβ42-induced FXII activation (lane 6 in [D-E]). FXIIa and PK western blots were normalized against TF. n = 6. Data are denoted as mean ± SEM. *P < .05, **P ≤ .01, ***P ≤ .001. P > .05 was not significant (n.s.).
Since activated PK (kallikrein) can feed back to activate FXII51 and 3E8 inhibited PK activation, we investigated whether 3E8 affected FXII activation. We found that 3E8 dose-dependently inhibited FXII activation (Figure 3D-E). These results suggest that 3E8 anti-HK antibody dose-dependently prevents both PK and FXII activation. Since our immunoprecipitation analyses showed that 3E8 only binds HK but not FXII or PK in human plasma (Figure 1B), the effects of 3E8 on FXII or PK activation are through its direct binding to HK, not FXII or PK.
3E8 has the same effect on Aβ42-induced FXII activation as does HK deficiency in human plasma
We compared the effect of 3E8 anti-HK antibody with that of HK deficiency (using KN-DF human plasma) on Aβ42-induced FXII activation in human plasma. In NHP, Aβ42 induced FXII activation and HK cleavage, and 3E8 completely blocked HK cleavage36 (Figures 1,-,3). Although significant, 3E8 did not completely prevent FXII activation (Figures 3D-E and 4). Using KN-DF human plasma, we found that Aβ42 induced a small but significant amount of FXII activation (lane 6 of Figure 4A-B). 3E8 did not have a significant effect on Aβ42-induced FXII activation in KN-DF plasma (lanes 6 vs 7 in Figure 4A-B). The addition of 3E8 to normal plasma and the overall lack of HK in KN-DF plasma have similar inhibitory effects on Aβ42-induced FXII activation (lanes 3 vs 6 in Figure 4A-B). These results indicate that HK plays an important role in Aβ42-induced FXII activation and that HK participates in contact system activation primarily through the 20-residue region of D6. Therefore, an antibody blocking this region, like 3E8, is effective in inhibiting contact system activation.
![3E8 anti-HK antibody has the same effects on Aβ42-induced FXII activation as does the absence of HK in human plasma. NHP or KN-DF human plasma was incubated with buffer, 3E8 (0.5 μM), or control IgG (0.5 μM) at 37°C for 20 minutes. Then Aβ42 (5 μM) was added and incubated at 37°C for 2 hours. Western blotting analyses were performed with antibodies against FXII/FXIIa, HK, and TF. In the absence of 3E8, Aβ42 induced FXII activation and HK cleavage in normal plasma (lane 2 in [A-B]). It also induced a small but significant activation of FXII in KN-DF human plasma (lane 6 in [A-B]). 3E8 blocked FXII activation in normal plasma but had no significant effect on FXII activation in KN-DF plasma (lane 7 in [A-B]). Control IgG did not affect Aβ42-induced FXII or HK changes. FXIIa and HK signals were normalized to TF. n = 6. Data are denoted as mean ± SEM. *P < .05, ***P ≤ .001. P > .05 was not significant (n.s.).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/10/10.1182_bloodadvances.2021006612/3/m_advancesadv2021006612f4.png?Expires=1765894666&Signature=ZUb91fwazAfe6D0XohpouydA0lF62iUf6dHLnjtwBEbhMkXeWtXWfS3kg2OfzlOorZout3G4pjW2P34wbXPDFKi9XhFdmWQSgts~3A5Ffeh0k3dZlHapLtLlyxARsYEqKteMgyiaV-LbBHx4K3XCaBZZFL7CbkprV-TNAvTsrv24S20-uacFQOd2PNg0km3xwSU~atSIKvWfvH5CUhdOggLZvUArBaNQAR85rdqHX-YqGU-A7Wwat~cHYgH20S9LT83RUvOL4Xvp5XPA8z9oOl-9oYiAW3IX1fEJ6-wP9GSoPSS0GDRizcFhjy4RSQqml2nuC6-xew2xyfS~ljXJcg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
3E8 anti-HK antibody has the same effects on Aβ42-induced FXII activation as does the absence of HK in human plasma. NHP or KN-DF human plasma was incubated with buffer, 3E8 (0.5 μM), or control IgG (0.5 μM) at 37°C for 20 minutes. Then Aβ42 (5 μM) was added and incubated at 37°C for 2 hours. Western blotting analyses were performed with antibodies against FXII/FXIIa, HK, and TF. In the absence of 3E8, Aβ42 induced FXII activation and HK cleavage in normal plasma (lane 2 in [A-B]). It also induced a small but significant activation of FXII in KN-DF human plasma (lane 6 in [A-B]). 3E8 blocked FXII activation in normal plasma but had no significant effect on FXII activation in KN-DF plasma (lane 7 in [A-B]). Control IgG did not affect Aβ42-induced FXII or HK changes. FXIIa and HK signals were normalized to TF. n = 6. Data are denoted as mean ± SEM. *P < .05, ***P ≤ .001. P > .05 was not significant (n.s.).
3E8 anti-HK antibody has the same effects on Aβ42-induced FXII activation as does the absence of HK in human plasma. NHP or KN-DF human plasma was incubated with buffer, 3E8 (0.5 μM), or control IgG (0.5 μM) at 37°C for 20 minutes. Then Aβ42 (5 μM) was added and incubated at 37°C for 2 hours. Western blotting analyses were performed with antibodies against FXII/FXIIa, HK, and TF. In the absence of 3E8, Aβ42 induced FXII activation and HK cleavage in normal plasma (lane 2 in [A-B]). It also induced a small but significant activation of FXII in KN-DF human plasma (lane 6 in [A-B]). 3E8 blocked FXII activation in normal plasma but had no significant effect on FXII activation in KN-DF plasma (lane 7 in [A-B]). Control IgG did not affect Aβ42-induced FXII or HK changes. FXIIa and HK signals were normalized to TF. n = 6. Data are denoted as mean ± SEM. *P < .05, ***P ≤ .001. P > .05 was not significant (n.s.).
3E8 delays intrinsic coagulation and prevents Aβ42-induced intrinsic coagulation
FXI plays an important role in the activation of the intrinsic coagulation pathway.52 Since 3E8 and FXI both bind D6 of HK, 3E8 might interfere with FXI binding and impact intrinsic coagulation. To analyze whether 3E8 affects intrinsic coagulation, we measured aPTT.53 After incubating human plasma with various concentrations of 3E8, we induced intrinsic coagulation. While a very low dose of 3E8 (0.05 μM) did not impact aPTT, a higher dose (0.15 μM) significantly delayed aPTT (Figure 5A). We also analyzed the effect of 3E8 on the extrinsic coagulation pathway by measuring PT.53 We found that 3E8 did not impact the PT (Figure 5B), indicating that 3E8 delays intrinsic, but not extrinsic, coagulation.

The 3E8 anti-HK antibody delays aPTT but not PT and prevents Aβ42-induced intrinsic coagulation in human plasma. (A) The 3E8 HK antibody delayed aPTT. Citrated human plasma was incubated with buffer, 3E8 (0.05 μM, 0.15 μM, 0.75 μM, 3.75 μM), or control IgG (3.75 μM) at 37°C for 20 minutes. Intrinsic coagulation was initiated by adding aPTT reagent and CaCl2 solution. Absorbance readings were recorded every 5 seconds at 350 nm, and aPTT was determined. 3E8 did not significantly affect aPTT at 0.05 μM, but it significantly delayed the aPTT at 0.15 μM. However, higher amounts of 3E8 did not cause further delay. Control IgG did not affect aPTT. (B) The 3E8 HK antibody did not affect PT. Citrated human plasma was incubated with buffer, 3E8 (3.75 μM), or control IgG (3.75 μM) at 37°C for 20 minutes. The extrinsic coagulation pathway was initiated by adding tissue factor with CaCl2. Absorbance readings were recorded every 5 seconds at 350 nm, and PT was determined. (C) 3E8 normalized Aβ42-induced intrinsic coagulation in human plasma. Citrated human plasma was incubated with buffer, 3E8 (0.025 μM, 0.075 μM, 0.25 μM), or control IgG (0.25 μM) at 37°C for 20 minutes. Aβ42 (4 μM) with CaCl2 was added to induce coagulation. 3E8 did not significantly affect Aβ42-induced coagulation at 0.025 μM, while 3E8 corrected Aβ42-induced coagulation at 0.075 μM and 0.25 μM. However, there was no significant difference between 0.075 μM and 0.25 μM 3E8 antibodies. Control IgG did not affect Aβ42-induced coagulation. n = 6. Data are denoted as mean ± SEM. **P ≤ .01, ***P ≤ .001. P > .05 was not significant (n.s.).
The 3E8 anti-HK antibody delays aPTT but not PT and prevents Aβ42-induced intrinsic coagulation in human plasma. (A) The 3E8 HK antibody delayed aPTT. Citrated human plasma was incubated with buffer, 3E8 (0.05 μM, 0.15 μM, 0.75 μM, 3.75 μM), or control IgG (3.75 μM) at 37°C for 20 minutes. Intrinsic coagulation was initiated by adding aPTT reagent and CaCl2 solution. Absorbance readings were recorded every 5 seconds at 350 nm, and aPTT was determined. 3E8 did not significantly affect aPTT at 0.05 μM, but it significantly delayed the aPTT at 0.15 μM. However, higher amounts of 3E8 did not cause further delay. Control IgG did not affect aPTT. (B) The 3E8 HK antibody did not affect PT. Citrated human plasma was incubated with buffer, 3E8 (3.75 μM), or control IgG (3.75 μM) at 37°C for 20 minutes. The extrinsic coagulation pathway was initiated by adding tissue factor with CaCl2. Absorbance readings were recorded every 5 seconds at 350 nm, and PT was determined. (C) 3E8 normalized Aβ42-induced intrinsic coagulation in human plasma. Citrated human plasma was incubated with buffer, 3E8 (0.025 μM, 0.075 μM, 0.25 μM), or control IgG (0.25 μM) at 37°C for 20 minutes. Aβ42 (4 μM) with CaCl2 was added to induce coagulation. 3E8 did not significantly affect Aβ42-induced coagulation at 0.025 μM, while 3E8 corrected Aβ42-induced coagulation at 0.075 μM and 0.25 μM. However, there was no significant difference between 0.075 μM and 0.25 μM 3E8 antibodies. Control IgG did not affect Aβ42-induced coagulation. n = 6. Data are denoted as mean ± SEM. **P ≤ .01, ***P ≤ .001. P > .05 was not significant (n.s.).
Aβ42 induces thrombin generation32 and dramatically accelerates clotting in normal plasma (Figure 5C). To analyze whether 3E8 anti-HK antibody could prevent Aβ42-induced clotting, we incubated human plasma with various concentrations of 3E8 and then Aβ42 before adding calcium to initiate coagulation. 3E8 (75 nM) abolished the effects of Aβ42-induced accelerated clotting and normalized clotting (Figure 5C).
Since FXI can also be activated by thrombin,54 we used a purified protein system to examine whether 3E8 could affect thrombin-induced FXI activation. Our results showed that the 3E8 anti-HK antibody did not inhibit FXI activation by thrombin (supplemental Figure 1).
Aβ42 binding to both HK and FXII, and PK and FXI binding to HK play important roles in Aβ42-induced plasma contact system activation
We analyzed the interaction between Aβ42 and other contact system proteins in NHP using biotinylated-Aβ42 (B-Aβ42). Molecules were pulled down and analyzed by nonreducing SDS-PAGE/western blots. The specificity of the antibodies used was validated using purified proteins (lanes 1-4, Figure 6A). B-Aβ42 pulled down FXII/FXIIa, cHK, some FXI, and some PK (Lane 7, Figure 6A). Since the contact system was activated by Aβ42 during the pulldown process: FXII was dramatically activated; HK was cleaved; PK and FXI were decreased due to their activation to PKa and actived FXI (FXIa), respectively, which are not detectable by these antibodies (compare lanes 5-7, Figure 6A). Most FXIIa was pulled down by Aβ42, as FXIIa was not detected in the supernatant after B-Aβ42 pulldown. Furthermore, cHK was pulled down by Aβ42 (lanes 5-7, Figure 6A). These results show that activated FXII and cleaved HK bind to Aβ42. TF, a noncontact system plasma protein used for normalization, was not pulled down by B-Aβ42, indicating the specificity of the pulldown assay.
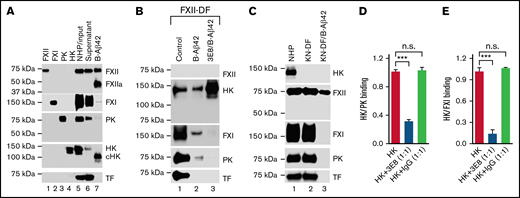
Aβ42 binding to both HK and FXII, and PK and FXI binding to HK are involved in Aβ42-induced plasma contact system activation. (A) Aβ42 pulls down FXII, cHK, FXI, and PK from pooled NHP. NHP was incubated with biotinylated Aβ42 (B-Aβ42). The B-Aβ42/bound protein complexes were pulled down by Dynabeads M-280 Streptavidin, eluted with SDS sample buffer, and analyzed by western blot. Purified human FXII, FXI, PK, and HK were used to validate the specificity of the antibodies for each protein (lanes 1-4). Each protein was detected in the NHP input (lane 5) and the supernatant after B-Aβ42 pulldown (lane 6). B-Aβ42 pulled down FXII and FXIIa (activated by Aβ42-induced contact system activation during the pulldown process), cHK (HK was cleaved due to contact system activation, so most, if not all, cHK was pulled down by Aβ42), FXI, and some PK (lane 7). During the pulldown process, B-Aβ42 activated the contact system leading to FXII activation, decreased PK (since it was activated to PKa, which was not detectable by this antibody), FXI molecular size shift, and HK cleavage (compare lanes 5-7). TF was not pulled down by B-Aβ42. (B) 3E8 anti-HK antibody blocked PK and FXI, but not HK, pulldown by B-Aβ42. Since the contact system was activated during the pulldown process, we used FXII-DF human plasma to avoid B-Aβ42-induced contact system activation. All proteins except for FXII were detected in FXII-DF plasma (lane 1). B-Aβ42 pulled down HK, FXI, and PK, but not TF (lane 2). 3E8 blocked FXI and PK pulldown by B-Aβ42, but not HK (lane 3). (C) HK deficiency in KN-DF plasma prevented FXI and PK pulldown by B-Aβ42. All proteins except HK were detected in KN-DF plasma, and protein levels were similar between PNP and KN-DF plasma (lanes 1 vs 2). B-Aβ42 pulled down FXII, but neither FXI nor PK from KN-DF plasma (lane 3). (D,E) Plates were coated with PK (D) or FXI (E) and then incubated with HK in the presence or absence of 3E8 anti-HK antibody. In the absence of 3E8, HK binds to PK (D) or FXI (E). In the presence of 3E8, binding is blocked between HK and PK (D) or FXI (E). The control IgG did not have an effect on HK binding to PK or FXI. The experiments were performed in triplicate and repeated 3 times. Data are denoted as mean ± SEM. ***P ≤ .001. P > .05 was not significant (n.s.).
Aβ42 binding to both HK and FXII, and PK and FXI binding to HK are involved in Aβ42-induced plasma contact system activation. (A) Aβ42 pulls down FXII, cHK, FXI, and PK from pooled NHP. NHP was incubated with biotinylated Aβ42 (B-Aβ42). The B-Aβ42/bound protein complexes were pulled down by Dynabeads M-280 Streptavidin, eluted with SDS sample buffer, and analyzed by western blot. Purified human FXII, FXI, PK, and HK were used to validate the specificity of the antibodies for each protein (lanes 1-4). Each protein was detected in the NHP input (lane 5) and the supernatant after B-Aβ42 pulldown (lane 6). B-Aβ42 pulled down FXII and FXIIa (activated by Aβ42-induced contact system activation during the pulldown process), cHK (HK was cleaved due to contact system activation, so most, if not all, cHK was pulled down by Aβ42), FXI, and some PK (lane 7). During the pulldown process, B-Aβ42 activated the contact system leading to FXII activation, decreased PK (since it was activated to PKa, which was not detectable by this antibody), FXI molecular size shift, and HK cleavage (compare lanes 5-7). TF was not pulled down by B-Aβ42. (B) 3E8 anti-HK antibody blocked PK and FXI, but not HK, pulldown by B-Aβ42. Since the contact system was activated during the pulldown process, we used FXII-DF human plasma to avoid B-Aβ42-induced contact system activation. All proteins except for FXII were detected in FXII-DF plasma (lane 1). B-Aβ42 pulled down HK, FXI, and PK, but not TF (lane 2). 3E8 blocked FXI and PK pulldown by B-Aβ42, but not HK (lane 3). (C) HK deficiency in KN-DF plasma prevented FXI and PK pulldown by B-Aβ42. All proteins except HK were detected in KN-DF plasma, and protein levels were similar between PNP and KN-DF plasma (lanes 1 vs 2). B-Aβ42 pulled down FXII, but neither FXI nor PK from KN-DF plasma (lane 3). (D,E) Plates were coated with PK (D) or FXI (E) and then incubated with HK in the presence or absence of 3E8 anti-HK antibody. In the absence of 3E8, HK binds to PK (D) or FXI (E). In the presence of 3E8, binding is blocked between HK and PK (D) or FXI (E). The control IgG did not have an effect on HK binding to PK or FXI. The experiments were performed in triplicate and repeated 3 times. Data are denoted as mean ± SEM. ***P ≤ .001. P > .05 was not significant (n.s.).
Since the pulldown process with B-Aβ42 induced contact system activation, we used FXII-deficient human plasma (FXII-DF) for further studies. All proteins except FXII were detected in FXII-DF (lane 1, Figure 6B). B-Aβ42 pulled down HK, FXI, and PK, but not TF (lane 2, Figure 6B). 3E8 blocked FXI and PK pulldown by B-Aβ42, but not HK (lanes 2 vs 3, Figure 6B), indicating FXI and PK pulldown by B-Aβ42 depends on their binding to HK. 3E8 blocked FXI and PK binding to HK, therefore preventing FXI and PK pulldown. FXII-deficiency prevented Aβ42-induced HK cleavage as cHK was not detected (Figure 6A vs Figure 6B), indicating that FXII is necessary for Aβ42-induced contact system activation.
To further analyze the role of HK in the interaction of FXI/PK with Aβ42, KN-DF plasma was used. All proteins analyzed except HK were detected in KN-DF plasma, and the protein levels were similar between NHP and KN-DF plasma (lanes 1 and 2, Figure 6C). B-Aβ42 pulled down FXII but neither FXI nor PK from KN-DF plasma (lane 3, Figure 6C). This result further shows that FXI and PK pulled down by Aβ42 depends on HK; in the absence of HK, Aβ42 did not pull down FXI and PK. Even though Aβ42 induced FXII activation and pulled down FXIIa in NHP (lane 7, Figure 6A), it did not pull down FXIIa in KN-DF plasma (lane 3, Figure 6C) as there was likely too little FXIIa formed to be pulled down (Figure 4). This result indicates that Aβ42 did not induce FXII activation in KN-DF plasma. Therefore, Aβ42 binding to both FXII and HK is critical for Aβ42-mediated contact system activation.
To further investigate the role of 3E8 anti-HK antibody in FXI and PK binding to HK, we used an ELISA-based binding assay. Plates were coated with PK (Figure 6D) or FXI (Figure 6E) and then incubated with HK in the presence or absence of 3E8. In the absence of 3E8, HK bound to PK (Figure 6D) or FXI (Figure 6E). In the presence of 3E8, HK binding to PK (Figure 6D) or FXI (Figure 6D) was blocked. Control IgG did not influence HK binding to PK or FXI. These results directly show that 3E8 blocked PK or FXI binding to HK.
3E8 anti-HK antibody disassembles PK/HK and FXI/HK complexes in NHP in the absence of a contact system activator ex vivo
Since PK and FXI circulate in the blood as complexes with HK, we hypothesized that 3E8 could disassemble PK/HK or FXI/HK complexes in NHP in the absence of contact system activators. To test this possibility, we incubated NHP with 3E8 anti-HK antibody, then pulled down HK and its bound proteins with an anti-HK antibody that does not recognize or bind D6 (biotinylated-2B7, B-2B7).44 In the absence of 3E8, B-2B7 anti-HK antibody pulled down HK, FXI, and PK (lanes 1 and 3, Figure 7A). However, in the presence of 3E8, B-2B7 antibody pulled down HK but not FXI or PK (lane 2, Figure 7A,C,D). This result indicates that the 3E8 anti-HK antibody disassembled PK/HK and FXI/HK complexes in human plasma in the absence of contact system activation ex vivo. There was significantly more intact HK pulled down in the presence of 3E8 compared with in the absence of 3E8 (Figure 7A,B). Control IgG did not have an effect on the pulldown, indicating the specificity of the 3E8 antibody. Neither FXII nor albumin were pulled down, demonstrating the specificity of the B-2B7 antibody for HK and the lack of binding between FXII and HK in the absence of a contact system activator. Overall, this experiment shows that the 3E8 anti-HK antibody is capable of disassembling PK/HK and FXI/HK complexes in normal human plasma in the absence of contact system activation ex vivo.
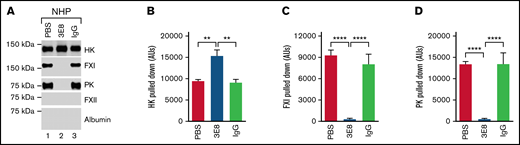
3E8 anti-HK antibody disassembles PK/HK and FXI/HK complexes in NHP in the absence of contact system activator ex vivo. NHP was incubated with PBS, 3E8, or hamster IgG at 37°C for 20 minutes. B-2B7 anti-HK antibody was added and incubated for an additional 20 minutes. Streptavidin was used to pull down B-2B7-bound proteins. (A) Representative western blot results of the pulldown experiments. While HK was pulled down in all samples, FXI and PK were only pulled down in PBS- or IgG-treated plasma (lanes 1 and 3 vs lane 2). (B) 3E8 anti-HK antibody pulled down significantly more intact HK than PBS/IgG samples. (C-D) FXI and PK were pulled down from PBS- and hamster IgG-treated, but not 3E8-treated NHP, indicating that 3E8 anti-HK antibody is able to disassemble PK/HK and FXI/HK complexes in normal human plasma in the absence of a contact system activator ex vivo. FXII and human albumin were not pulled down in any of the samples. n = 8 per group. Data are denoted as mean ± SEM. **P ≤ .01, ****P ≤ .0001.
3E8 anti-HK antibody disassembles PK/HK and FXI/HK complexes in NHP in the absence of contact system activator ex vivo. NHP was incubated with PBS, 3E8, or hamster IgG at 37°C for 20 minutes. B-2B7 anti-HK antibody was added and incubated for an additional 20 minutes. Streptavidin was used to pull down B-2B7-bound proteins. (A) Representative western blot results of the pulldown experiments. While HK was pulled down in all samples, FXI and PK were only pulled down in PBS- or IgG-treated plasma (lanes 1 and 3 vs lane 2). (B) 3E8 anti-HK antibody pulled down significantly more intact HK than PBS/IgG samples. (C-D) FXI and PK were pulled down from PBS- and hamster IgG-treated, but not 3E8-treated NHP, indicating that 3E8 anti-HK antibody is able to disassemble PK/HK and FXI/HK complexes in normal human plasma in the absence of a contact system activator ex vivo. FXII and human albumin were not pulled down in any of the samples. n = 8 per group. Data are denoted as mean ± SEM. **P ≤ .01, ****P ≤ .0001.
Discussion
HK is a nonenzymatic cofactor for the optimal activation of the plasma contact system. HK mostly circulates in the blood complexed with PK or FXI.55,56 FXII binds directly to negatively charged surfaces, which can lead to autoactivation, whereas HK may be necessary for optimal activation of PK and FXI by surface-bound activated FXIIa and for positive feedback activation of FXII by PKa.57,58 Plasma deficient in HK has a markedly prolonged aPTT ex vivo.59-62
Human HK consists of 6 domains (designated D1 to D6, respectively). Previous studies revealed that D6 of HK contains the PK and FXI binding sites, which partially overlap.47-50 This region is important for HK-dependent clotting activity of NHP and dextran sulfate sodium salt-induced activation of PK and HK cleavage.47 We focused on a smaller 20-amino acid region within the identified PK/FXI binding site in D6 of HK.47-49 The effect of 3E8 anti-HK antibody on Aβ42-induced HK cleavage is dose-dependently blocked by the HK-D6 peptide (Figure 2), indicating the importance of this region. Moreover, 3E8 inhibited FXII and PK activation and delayed aPTT in NHP. Our immunoprecipitation experiment in human plasma showed that 3E8 directly bound HK yet bound neither FXII nor PK, indicating the effects of 3E8 on contact system activation is through its binding to HK. Together, these results suggest the effects of 3E8 anti-HK antibody on Aβ42-induced contact system activation is through its binding to a 20-amino acid region of the D6 of HK, demonstrating that this small region of HK plays an important role in Aβ42-induced contact system activation.
Based on previous studies from other investigators47-50,55,56,58 and our present results, we hypothesize the following models: in normal plasma (supplemental Figure 2A), PK and FXI bind to HK and form complexes.50,56 Aβ42 binds to FXII and activates FXII to FXIIa, which initiates contact system activation. Aβ42 also binds to HK, which facilitates access of PK and FXII to Aβ42-bound FXIIa (Figure 6A). FXIIa activation of FXI (to FXIa) triggers the intrinsic coagulation pathway, leading to clot formation, whereas FXIIa activation of PK (to PKa/kallikrein) leads to the cleavage of HK and release of bradykinin from its precursor HK and subsequent binding to bradykinin receptors and initiation of inflammatory processes. PKa can positively feedback to activate FXII,51 and FXIIa can further activate the downstream pathways and amplify contact system activation. In the presence of 3E8 (supplemental Figure 2B), 3E8 binds to D6 of HK and blocks PK and FXI binding to HK (Figure 6B,D,E). Aβ42 can minimally activate FXII in the presence of 3E8 (Figure 4), but Aβ42-bound FXIIa does not have access to FXI and PK due to the binding of 3E8 and therefore cannot activate PK or FXI. Since PK is not activated, it cannot feedback to activate more FXII, and FXII remains minimally activated by Aβ42 (Figure 3D). The effect of KN-DF is similar to that of 3E8. In the absence of HK (supplemental Figure 2C), Aβ42 can bind to FXII and initiate limited FXII activation (Figure 4), but Aβ42-bound FXIIa does not have access to FXI and PK and therefore cannot activate PK nor FXI. Since PK is not activated, it cannot positively feedback to activate FXII, and FXII has very limited activation by Aβ42 (Figures 4 and 6C). Therefore, in the absence of HK, contact system activation is very limited by Aβ42 (Figure 4).
HK has important functions in many pathophysiological conditions.39,63-65 Kininogen-1 knockout mice are protected from ischemic neurodegeneration without an increase in infarct-associated hemorrhage.66 Therefore, HK is instrumental in pathologic thrombus formation and inflammation but dispensable for hemostasis.66 Bradykinin, which is released from HK, is involved in many pathologies in a variety of systems and organs.40,67-74 These studies suggest that targeting HK may be beneficial in many pathological conditions. Our study showed that 3E8, an antibody targeting HK, not only blocks bradykinin release36 and subsequent inflammatory activity, but it also inhibits FXI and PK activation, prevents FXII feedback activation of FXII, and prevents intrinsic coagulation ex vivo. Furthermore, 3E8 can disassemble HK/PK and HK/FXI complexes in the absence of a contact system activator. Our studies may provide important information for novel therapeutic and prophylactic strategies for contact system-related pathological conditions, such as Alzheimer’s disease and hereditary angioedema.
Acknowledgments
The authors thank members of the Strickland laboratory for helpful discussions.
This work was supported by the National Institutes of Health (NIH) grants NS102721 and AG069987, Cure Alzheimer’s Fund, Alzheimer’s Association, Robertson Therapeutic Development Fund, Samuel Newhouse Foundation, Mr. John Herrmann, and Rudin Family Foundation; also supported by UL1TR001866 from the National Center for Advancing Translational Sciences, NIH Clinical and Translational Science Award program.
Authorship
Contribution: Z-L.C. designed the study, performed experiments, analyzed data, and wrote the manuscript; P.K.S. and K.H. performed experiments and analyzed data; S.S. helped design the study and analyze data; and E.H.N. designed the study, participated in data analysis, and wrote the manuscript.
Conflict-of-interest disclosure: The 3E8 and 2B7 anti-HK antibodies have been licensed to Millipore Sigma. The authors declare no other competing financial interests.
Correspondence: Erin H. Norris, The Rockefeller University, 1230 York Avenue, Box 169, New York, NY 10065; e-mail: enorris@rockefeller.edu.

