

Key Points
Adult patients with acute lymphoblastic leukemia with low MRD positivity at week 16 form an intermediate-risk group.
NGS improves the risk assignment of patients with MolNE MRD.

Abstract
Persistence of minimal residual disease (MRD) after induction/consolidation therapy in acute lymphoblastic leukemia is the leading cause of relapse. The GMALL 07/2003 study used MRD detection by real-time quantitative polymerase chain reaction of clonal immune gene rearrangements with 1 × 10−4 as discriminating cutoff: levels ≥1 × 10−4 define molecular failure and MRD-negativity with an assay sensitivity of at least 1 × 10−4 defining complete molecular response. The clinical relevance of MRD results not fitting into these categories is unclear and termed “molecular not evaluable” (MolNE) toward MRD-based treatment decisions. Within the GMALL 07/03 study, 1019 consecutive bone marrow samples after first consolidation were evaluated for MRD. Patients with complete molecular response had significantly better outcome (5-year overall survival [OS] = 85% ± 2%, n = 603; 5-year disease-free survival [DFS] = 73% ± 2%, n = 599) compared with patients with molecular failure (5-year OS = 40% ± 3%, n = 238; 5-year DFS = 29% ± 3%, n = 208), with patients with MolNE in between (5-year OS = 66% ± 4%; 5-year DFS = 52% ± 4%, n = 178). Of MolNE samples reanalyzed using next-generation sequencing (NGS), patients with undetectable NGS-MRD (n = 44; 5-year OS = 88% ± 5%, 5-year DFS = 70% ± 7%) had significantly better outcome than those with positive NGS-MRD (n = 42; 5-year OS = 37% ± 8%; 5-year DFS = 33% ± 8%). MolNE MRD results not just are borderline values with questionable relevance but also form an intermediate-risk group, assignment of which can be further improved by NGS.
Introduction
Minimal residual disease (MRD) detection is standard practice in the treatment of adult patients with acute lymphoblastic leukemia (ALL) across Europe, with 73% of patients being tested for MRD in first complete remission.1 With standard chemotherapy regimens, ∼90% of patients achieve hematological remission, defined as <5% of blasts in bone marrow based on morphologic assessment and resolution of extramedullary involvement. However, with the use of modern technologies, such as multiparameter flow cytometry and real-time quantitative polymerase chain reaction (RQ-PCR), 30% to 50% of patients have persistent MRD below the detection level of routine microscopy, which is the main cause of subsequent relapse.1-4
Detectable MRD after induction/consolidation therapy has been associated with poorer disease-free (DFS) and overall survival (OS) across various therapies, disease subtypes, detection methods, and time points.5,6 MRD is therefore widely used to stratify patients into risk groups, to adjust the intensity of chemotherapy, to decide on the addition of targeted therapies, for timely recognition of impending relapse, and as an early measure of disease response in clinical trials.
Most studies employ 1 × 10−4 as a cutoff for defining MRD persistence/relapse detected by allele-specific RQ-PCR of clonal immunoglobulin and T-cell receptor (TR) gene rearrangements.7 Also, within the German Multicenter ALL trial (GMALL) 07/2003 and the subsequent observational study, RQ-PCR was prospectively used for MRD detection and a cutoff of 1 × 10−4 at treatment week 10 (w+10), and w+16 was used to identify patients with molecular failure (MolFail).4 This approach minimizes the risk of obtaining false-positive RQ-PCR results owing to a nonspecific amplification of healthy background lymphoid cells in regenerating bone marrow.8,9 Although MRD positivity <1 × 10−4 is assumed to reflect real low-level disease in most cases, its prognostic relevance has not yet been defined in adult ALL. Even though the EuroMRD group developed precise guidelines for clinical situations where false positivity or false negativity have to be prevented, nonspecific amplification of background immunoglobulin/TR rearrangements might be mixed up with real low-level positivity below quantitative range.10
Here, we analyzed the prognostic relevance of MRD results not fitting to the categories MolFail (quantifiable MRD ≥1 × 10−4) or complete molecular response (MolCR: MRD negativity with an assay sensitivity of at least 1 × 10−4) after consolidation treatment I at week 16 (w+16) of the GMALL 07/2003 protocol, including Ph− ALL patients aged 15 to 55 years. Where possible, we retrospectively reanalyzed these samples with next-generation sequencing (NGS)-based immunoglobulin/TR assays to evaluate whether the reportedly better specificity of NGS may enhance prognostication.
Methods
The entire analyzed cohort contained 1019 high-risk and standard-risk consecutive patients from the GMALL 07/2003 study with Ph− ALL, whose MRD in bone marrow aspirate at w+16 was analyzed at the Unit for Hematological Diagnostics in Kiel by the EuroMRD-based immunoglobulin/TR RQ-PCR. Of the samples, 178 did not fall into the MolCR (MRD negativity with sensitivity at least 10−4) or the MolFail (MRD ≥ 10−4) groups because they were either MRD− with an insufficient assay sensitivity, MRD+ below quantitative range,10 or MRD+ below 1 × 10−4. Together, these patients were classified as molecular not evaluable (MolNE), and their clinical characteristics are summarized in Table 1. Risk stratification on the GMALL 07/2003 and its therapeutic consequences are summarized in the supplemental material. The study was approved by the Institutional Review Board of the Christian Albrechts-University in Kiel and performed in accordance with the Declaration of Helsinki.
Characteristics of the MolNE patient group
| Patients (N = 178) | ||
|---|---|---|
| Characteristics | No. | % |
| Age, y | ||
| Median (range) | 29 (15-64) | |
| ≤35 y | 112 | 63 |
| >5 y | 66 | 37 |
| Risk stratification | ||
| Standard risk | 131 | 74 |
| High risk | 47 | 26 |
| Immunophenotype | ||
| C-/pre-B-ALL | 111 | 62 |
| Pro-B-ALL | 11 | 6 |
| Early-T-ALL | 7 | 4 |
| Mature T-ALL | 7 | 4 |
| Thymic-T-ALL | 42 | 24 |
| White blood cell count, /µL | ||
| Median (range) | 11400 (400-463900) | |
| Patients (N = 178) | ||
|---|---|---|
| Characteristics | No. | % |
| Age, y | ||
| Median (range) | 29 (15-64) | |
| ≤35 y | 112 | 63 |
| >5 y | 66 | 37 |
| Risk stratification | ||
| Standard risk | 131 | 74 |
| High risk | 47 | 26 |
| Immunophenotype | ||
| C-/pre-B-ALL | 111 | 62 |
| Pro-B-ALL | 11 | 6 |
| Early-T-ALL | 7 | 4 |
| Mature T-ALL | 7 | 4 |
| Thymic-T-ALL | 42 | 24 |
| White blood cell count, /µL | ||
| Median (range) | 11400 (400-463900) | |
B-ALL, B-cell acute lymphoblastic leukemia; T-ALL, T-cell acute lymphoblastic leukemia.
For all patients, sequences of dominant immunoglobulin/TR rearrangements at the time of diagnosis, obtained by the classical low-throughput analysis employing multiplex BIOMED-2 PCRs11 and Sanger sequencing, were available. In 96 patients with diagnostic DNA available, EuroClonality NGS-based marker screening employing IGH-VJ-FR1 and TRB-VJ primers12 was performed to confirm the results of the routine low-throughput marker screening and as the basis for the NGS-based MRD quantification in w+16 samples. MRD at w+16 was quantified by NGS in 86 patients with available DNA with IGH-VJ-FR1 (60 rearrangements) and TRB-VJ (33 rearrangements) EuroClonality primers and 1-step PCR.12,13 For each patient, 3 replicates each containing 500 ng were analyzed, making the sensitivity of the assay comparable to RQ-PCR (1 × 10−5). For more details on NGS-based marker identification and MRD detection, please see the supplemental material.
Results and discussion
Of 1019 patients with Ph− acute lympholastic leukemia treated according to the GMALL07/2003 protocol, 603 (59%) were classified as MolCR according to the RQ-PCR MRD level at w+16, 238 (23%) were classified as MolFail, and 178 (17%) patients were classified as MolNE. Among patients with MolNE, 50 (28%) were MRD− with insufficient sensitivity, 4 (2%) had quantifiable MRD <1 × 10−4, and 124 (70%) had not-quantifiable MRD (with 57 of the respective RQ-PCR assays reaching a quantitative range of at least 1 × 10−4). The MolNE group of patients showed an intermediate prognosis (5-year OS = 66% ± 4%, 5-year DFS = 52% ± 4%, 5-year remission duration [RD] = 59% ± 4%) as compared with MolCR (5-year OS = 85% ± 2%, 5-year DFS = 73% ± 2%, 5-year RD = 80% ± 2%) and patients with MolFail (5-year OS = 40% ± 3%, 5-year DFS = 29% ± 3%, 5-year RD = 37% ± 4%; Figure 1A). This is in line with the results from a study on 304 adult patients with Ph− ALL, which showed that DFS of patients with postinduction MRD < 10−4 (52%) clustered between patients in complete molecular remission (63%), patients with MRD ranging from 10−4 to <10−3 (47%), and patients with MRD ≥ 10−3 (15%).14 Furthermore, a pediatric study on 455 children with B-ALL showed that patients with MRD levels between 1 × 10−4 and 1 × 10−5 at the end of remission induction therapy had a significantly higher risk of relapse than patients with lower or undetectable MRD.15
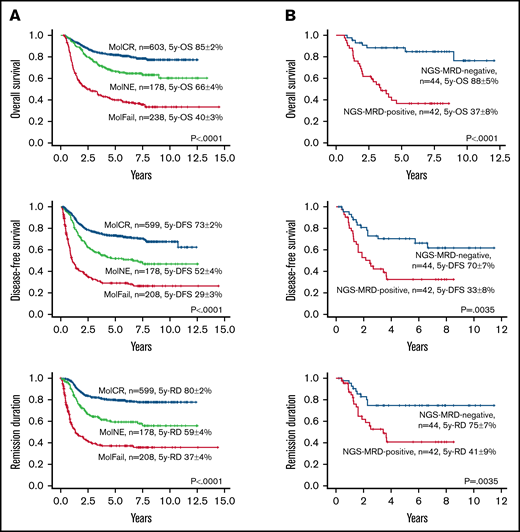
Prognostic impact of MRD levels at end of consolidation I (w+16), as shown by Kaplan-Meier estimates of OS, DFS, and RD. (A) OS, DFS, and RD in the total cohort of patients stratified by RQ-PCR as MolCR (MRD negativity with assays sensitivity of at least 10−4), MolFail (MRD positivity with a level of at least 10−4), and patients not fitting into these categories (MolNE). (B) OS, DFS, and RD according to NGS-MRD in patients who were MolNE by RQ-PCR. x-axis, years; y-axis, survival probability.
Prognostic impact of MRD levels at end of consolidation I (w+16), as shown by Kaplan-Meier estimates of OS, DFS, and RD. (A) OS, DFS, and RD in the total cohort of patients stratified by RQ-PCR as MolCR (MRD negativity with assays sensitivity of at least 10−4), MolFail (MRD positivity with a level of at least 10−4), and patients not fitting into these categories (MolNE). (B) OS, DFS, and RD according to NGS-MRD in patients who were MolNE by RQ-PCR. x-axis, years; y-axis, survival probability.
NGS-based MRD detection was performed to check RQ-PCR–based MRD results in w+16 samples of 86 patients of the MolNE group (67 patients with RQ-PCR+ MRD and 19 with RQ-PCR− MRD with insufficient sensitivity). The NGS assay detected MRD in 42/86 samples (49%), including 41/67 (61%) RQ-PCR+ and 1/19 (5%) RQ-PCR− with insufficient assay sensitivity. Of note, 26/67 (39%) of RQ-PCR+ MolNE samples were NGS-MRD−, suggesting a high rate of false positive MRD detection because of nonspecific amplification in this group. These data suggest that RQ-PCR negativity is generally confirmed by NGS and prognostically favorable even if the assay sensitivity does not formally reach the level of 1 × 10−4 according to EuroMRD criteria.10 RQ-PCR is the only method formally checking the sensitivity of each individual assay, whereas multiparameter flow cytometry and NGS only assume certain sensitivities based on input sample amount.
The 5-year OS and DFS of patients with NGS-MRD− (n = 44; 5-year OS = 88% ± 5%, 5-year DFS = 70% ± 7%, 5-year RD = 75% ± 7%) was significantly higher than for patients with NGS-MRD+ (n = 42; 5-year OS = 37% ± 8%, P < .0001; 5-year DFS = 33% ± 8%, P = .0035; 5-year RD = 41% ± 9%, P = .0035; Figure 1B). Outcome in patients with RQ-PCR− MolNE was excellent. The single NGS-MRD+/RQ-PCR− patient in this group died in complete remission after allogeneic stem cell transplantation. When patients with MolNE with negative RQ-PCR and insufficient assay sensitivity were excluded from the analysis, the difference between the OS of the 2 groups remained highly significant (NGS-MRD− patients: n = 26, 5-year OS = 84% ± 9%; NGS-MRD+ patients: n = 41, 5-year OS = 37% ± 8%, P = .0024; supplemental Figure 2). For more information on NGS-based marker screening and MRD detection results, please see the supplemental material.
In summary, we show that NGS improved the specificity of the MRD analysis and enabled a more precise risk prediction for patients with low-level nonquantifiable RQ-PCR MRD positivity. Although exact MRD quantification using immunoglobulin/TR NGS is still a challenge,13 distinction of MRD-positivity and MRD-negativity is possible and reliable, thanks to a more specific, nucleotide sequence–based readout avoiding false positive signals resulting from nonspecific amplification of background cells, as often observed in RQ-PCR.8,9,16 This limited specificity of RQ-PCR MRD assessment in this group of samples has been reported previously and represents a challenge for a reliable relapse risk prediction.8,9,16
Our results might seem to be in contrast with previous publications,17,18 mainly reporting false negative RQ-PCR MRD results. This illusive discrepancy is attributable to sample selection: whereas the other studies are performed on unselected samples, we focused on samples with low-level RQ-PCR MRD with a therefore higher probability of false positive RQ-PCR results. An NGS-MRD assessment using higher DNA amounts could potentially improve the sensitivity of the analysis and therefore the prognostication in the MolCR group, as suggested by others.19
Overall, NGS-based MRD analysis in ALL seems to be especially helpful in patients with RQ-PCR+ MolNE and may help to discriminate true MRD from false positivity in this considerable and clinically important group. The better specificity of NGS could potentially also improve the predictive value of MRD in the MolCR and MolFail groups; however, this is beyond the scope of this report. Still, further prospective studies are necessary to prove that NGS-MRD–based stratification can improve the outcome of patients with MolNE. Moreover, such studies could also help refine the definition of and assignment to the MolNE group and its 3 distinct subgroups (MRD− with insufficient sensitivity, quantifiable MRD <1 × 10−4, and not-quantifiable MRD), and eventually consolidate its significance and use beyond the GMALL study group.
Acknowledgments
The authors thank all the patients and physicians for their participation in the study. They also thank Maike Ipsen, Petra Chall, and Birgit Fricke for their excellent technical support and Henrik Knecht, Dietrich Herrmann, and Martin Schwarz for their help with NGS analysis. They also thank Regina Reutzel and Carina Fuchs, who served as GMALL administrative study coordinators.
Authorship
Contribution: J.K., N.G., and M.B. designed the study; M.K., J.K., H.T., N.D., C.D.B., B.S., J.B., N.A., K.W., N.G., J.B., K.N., S.R., A.V., and M.B. contributed and interpreted data; J.K. and H.T. performed and analyzed experiments; M.K., J.K., N.G., and M.B. designed and performed statistical analyses; C.D.B., N.G., and M.B. supervised the project; M.K. and M.B. drafted the first version of the manuscript; and all authors revised and approved the final version of the manuscript.
Conflict-of-interest disclosure: N.A. received personal fees from Gilead, MSD Sharp & Dohme GmbH, Pfizer and Amgen (advisory board), grants from Pfizer, and travel grants from Gilead, MSD Sharp & Dohme GmbH, Pfizer, and Amgen, all outside of the submitted work. K.N. received speaker’s honoraria from Jazz und BSH Medical, and travel support from Celgene. B.S. received support for attending meetings and travel from Jazz, Abbvie, Pfizer, Daiichi Sankyo, and Amgen, all outside of the submitted work. A.V. received personal fees from Amgen (advisory board) and Roche Pharma AG (advisory board, honoraria), Novartis (advisory board), Kite/Gilead (advisory board), and BMS (advisory board), all outside of the submitted work. C.D.B. received personal fees from BMS, Amgen, Novartis, and Jazz (advisory board) all outside of the submitted work. N.G. received speaker honoraria, travel support or advisory board fees from Amgen, Celgene, Gilead, Novartis, Pfizer, Jazz Pharmaceuticals, Incyte, Cellestia, Erytech, and Morphosys and research support (institution) from Amgen, Pfizer, Novartis, Shire/Servier, Jazz Pharmaceuticals, and Incyte. M.B. received personal fees from Incyte (advisory board) and Roche Pharma AG, financial support for reference diagnostics from Affimed and Regeneron, grants and personal fees from Amgen (advisory board, speakers bureau, travel support), and personal fees from Janssen (speakers bureau), all outside of the submitted work. The remaining authors declare no competing financial interests.
Correspondence: Monika Brüggemann, Unit for Hematological Diagnostics, Medical Department II, University Medical Center Schleswig-Holstein, Campus Kiel, Langer Segen 8-10, 24105 Kiel, Germany; e-mail: m.brueggemann@med2.uni-kiel.de.

