

Key Points
Fixed-duration chemotherapy-free treatment (obinutuzumab + idelalisib) is effective in R/R WM, although toxicity is a limitation.

Abstract
We present the results of a phase 2 study evaluating the combination of obinutuzumab + idelalisib in relapsed/refractory (R/R) Waldenström macroglobulinemia (WM). The goal was to determine the safety and efficacy of a fixed-duration chemotherapy-free treatment. During the induction phase, patients received idelalisib + obinutuzumab for 6 cycles, followed by a maintenance phase with idelalisib alone for ≤2 years. Forty-eight patients with R/R WM were treated with the induction combination, and 27 patients participated in the maintenance phase. The best responses, reached after a median of 6.5 months (interquartile range, 3.4-7.1; range, 2.6-22.1 months), were very good partial response in 5 patients, partial response in 27 patients, and minor response in 3 patients, leading to overall response rate and major response rate estimates of 71.4% (95% confidence interval [CI], 56.7-83.4) and 65.3% (95% CI, 50.4-78.3), respectively. With a median follow-up of 25.9 months, median progression-free survival was 25.4 months (95% CI, 15.7-29.0). Univariate analysis focusing on molecular screening found no significant impact of CXCR4 genotypes on responses and survivals but a deleterious impact of TP53 mutations on survival. Although there was no grade 5 toxicity, 26 patients were removed from the study because of side effects; the most frequent were neutropenia (9.4%), diarrhea (8.6%), and liver toxicity (9.3%). The combination of idelalisib + obinutuzumab is effective in R/R WM. Nonetheless, the apparent lack of impact of genotype on outcome could give new meaning to targeting of the phosphatidylinositol 3-kinase pathway in WM. This trial was registered at www.clinicaltrials.gov as #NCT02962401.
Introduction
Waldenström macroglobulinemia (WM) is characterized by the malignant accumulation of immunoglobulin M (IgM) that is caused by IgM-secreting lymphoplasmacytic lymphoma cells in bone marrow (BM).1 Since the initial development of treatment recommendations at the Second International Workshop on Waldenström Macroglobulinemia,2 treatment options for WM have evolved from immunochemotherapy to new compounds, such as Bruton tyrosine kinase (BTK) inhibitors, either alone or in combination with rituximab.3 The B-cell receptor (BCR) has been identified as a potential therapeutic target in many mature B-cell malignancies with distinct and/or heterogeneous physiopathology or clinical course. BCR-associated kinases, such as BTK or phosphatidylinositol 3-kinase δ (PI3Kδ), are critical signaling transducers of BCRs supporting the survival and growth of malignant B cells.4
Mutation of MYD88L265P was described by Treon and colleagues in 90% of WM patients; it is responsible for the proliferation and survival of WM cells by activating the NF-κB pathway via BTK.5,6 As a result, targeting of BCR kinases is emerging as a new treatment paradigm for numerous B-cell malignancies. Ibrutinib, a selective BTK inhibitor, has shown high activity and very promising results in patients with WM. Most chemotherapy-free regimen data have been reported in relapsing or refractory (R/R) patients treated with BTK inhibitors, alone or in association with rituximab.7-10 However, lower response rates and longer time to reach a response have been reported in MYD88WT and CXCR4MUT genotype patients treated with ibrutinib.7,11
Along with ibrutinib, idelalisib, an orally active selective PI3Kδ inhibitor, represents a new class of agents targeting signal transduction downstream from BCR in malignant B cells. MYD88L265P also promotes activation of the PI3K pathway constitutively activated in WM.12 In preclinical studies, idelalisib was shown to abrogate AKT activation, and it may target the BCR-controlled microenvironmental interaction of WM cells.13 Idelalisib has manifested antitumor activity in patients with previously treated indolent non-Hodgkin lymphomas.14 Compared with placebo + rituximab, the combination of idelalisib + rituximab appeared to shorten the period of lymphocytosis and improved progression-free survival (PFS), overall response rate (ORR), and overall survival (OS) in patients with relapsed chronic lymphocytic leukemia and clinically significant coexisting medical conditions, with an acceptable safety profile.15 However, idelalisib has been associated with significant and limiting adverse effects (AEs), particularly hepatic and infectious effects.16
Obinutuzumab, a glycoengineered type II anti-CD20 monoclonal antibody (mAb), has been described as being more effective than rituximab in low-grade lymphoid malignancies, including in unfit treatment-naive patients with chronic lymphocytic leukemia and in follicular lymphoma, resulting in longer PFS than with rituximab-based therapy.17,18 Use of a chemotherapy-free treatment, anti-CD20 mAb in combination with oral idelalisib, should allow some patients with WM, especially those who are medically unfit, to achieve a response and experience improved outcome and quality of life.
We present the results of a single-arm multicentric phase 2 study evaluating the combination of obinutuzumab + idelalisib in R/R WM.
Patients and methods
The trial was approved by an institutional review board of national ethical committees, Agence Nationale de Sécurité du Médicament et des Produits de Santé and Comité de Protection des Personnes. All patients provided written consent. This trial was registered at www.clinicalstrial.gov as #NCT02962401. Enrollment lasted from March 2017 until July 2018. Data updates were performed in April 2020.
Patients were eligible if they met the Second International Workshop on Waldenström's Macroglobulinemia diagnostic criteria1 and criteria to treat.19 Patients were relapsing (progressing >6 months after the last administration of first-line or subsequent treatment) or refractory (progressing ≤6 months after first-line or subsequent treatment) after 1 to 3 prior regimens/lines.
The primary end point was PFS, calculated from the date of inclusion to progression or death. Secondary end points included ORR, response rates (according to the VIth International Workshop on Waldenström's Macroglobulinemia criteria20 ) at months 8 and 24, OS, time to at least a minor response (MR), time to treatment failure (TTF), and time to new treatment. Secondary objectives were to assess safety and tolerability and the effect of MYD88 mutations (MYD88MUT) and CXCR4 mutations (CXCR4MUT) on response and PFS.
Measurable disease was required (ie, serum IgM ≥2 times the upper limit of normal [ULN]). Patients were required to meet the following pretreatment laboratory criteria: platelet count ≥75 × 109/L and absolute neutrophil count ≥1.5 × 109/L if not the result of BM infiltration; creatinine clearance ≥40 mL/min; total bilirubin ≤2 times the ULN, unless clearly related to the disease or Gilbert syndrome; serum alanine aminotransferase levels ≤2.5 times the ULN; aspartate aminotransferase ≤2.5 times the ULN; and Eastern Cooperative Oncology Group Performance Status ≤2. Patients with central nervous system involvement or with previous liver disease were excluded.
Serum IgM levels and complete blood cell counts were obtained at the beginning of each cycle for 6 cycles and then every 2 cycles thereafter. BM biopsies and computed tomography scans (if extramedullary disease was present at baseline) were repeated at cycles 6 and 24.
Drugs
During induction, idelalisib was given continuously (150 mg, twice a day orally) in association with IV obinutuzumab (100 mg on day 1, 900 mg on day 2, and 1000 mg on days 8 and 15 of cycle 1 and, thereafter, on day 1 of cycles 2 to 6 (28-day cycle). During the maintenance phase, idelalisib was given alone for ≤2 years.
Safety
Patients were closely monitored for infusion-related reactions. AEs were graded per Common Terminology Criteria for Adverse Events v.4.0. Idelalisib was held for absolute neutrophil count <0.5 × 109/L, grade ≥2 diarrhea, pneumonitis (of any grade), or grade ≥3 liver toxicity. Full-dose retreatment was permitted after toxicity recovery from the first drug hold; for subsequent events, reduction to 100 mg, twice a day, followed by discontinuance were required. Filgrastim or transfusion support was permitted. Herpesvirus and Pneumocystis prophylaxis were mandatory. All patients were tested before the initiation of treatment for antibody-response status (IgG and IgM) against cytomegalovirus (CMV) antigens (serology). CMV pp65 antigen or polymerase chain reaction (PCR) testing was obtained at screening in CMV-seropositive patients, followed by every month during the induction phase and every 2 months during the maintenance phase.
Gene mutation analysis
Next-generation sequencing (NGS) was performed centrally on DNA extracted from cells following Ficoll-Paque gradient centrifugation of BM samples collected at inclusion using the Ion PGM Torrent platform (Thermo Fisher Scientific). The custom gene panel included MYD88, CXCR4, TP53, BTK, and PLCG2. A mean coverage of 2000 to 2500× was obtained for all targeted regions, allowing analysis of subclonal variation with a limit of detection of 0.8%.21 In addition, droplet digital PCR was used to screen MYD88L265P and CXCR4S338X variants, with a limit of detection of 0.05%, according to the manufacturer’s recommendations (Bio-Rad).
Statistical analysis
The study aimed to assess whether PFS in advanced WM is improved by the combination of obinutuzumab + idelalisib compared with the combination applied in the usual therapy. The main chemotherapy-free approaches available when we designed the study were rituximab monotherapy or the combination of bortezomib, rituximab, and dexamethasone. We wished to demonstrate a median PFS ≥25 months compared with 15 months under the null hypothesis. With α = 0.05, power = 0.90, 24-month enrollment, and an assumed dropout rate at 0.010, the number of patients required was 50, based on a 1-sample log-rank test.22 All summary statistics are medians (interquartile range [IQR]) or percentages. Swimmer plots were plotted to display individual patient data over the trial. Analysis of efficacy used an intent-to-treat principle, based on all enrolled patients. PFS, OS, and TTF were estimated by the Kaplan-Meier method. Cumulative incidence of new treatment used deaths free of treatment change as competing risk. The safety population considered patients who received ≥1 dose of idelalisib. Data Safety Monitoring Board (DSMB) meetings were planned after the first 5 patients had received 3 cycles, after 25 enrolled patients, and after induction of treatment in 50 patients. All AEs and SAEs were submitted to the DSMB by the study statistician, independent of the sponsor and the investigators. Bayes estimation of the probability of grade ≥3 AEs was computed, using a β binomial model with noninformative uniform priors. All analyses were performed using R 3.5.1 (http://www.R-project.org). Two-sided P values ≤ .05 denoted statistical significance.
Results
Patients and disease characteristics
Fifty patients were enrolled across 21 sites. After 1 screen failure, 49 patients were actually included. Baseline characteristics of patients and mutations are listed in Table 1. No mutation in BTK or in PLCG2 was found in 3 patients previously treated with ibrutinib.
Baseline characteristics of enrolled patients (n = 49)
| Characteristics | Values |
|---|---|
| Age, y | 68.5 (50-83) |
| Males/females, n | 36/13 |
| ECOG PS, 0/1/2/unknown, n | 14/26/8/1 |
| Adenopathy/splenomegaly/hepatomegaly, n | 18/6/3 |
| White blood cells, ×109/L | 5.25 (4.1-7.3) |
| Hemoglobin, g/dL | 10 (8.9-11.1) |
| Platelets, ×109/L | 189 (94-2751) |
| Serum IgM, g/L | 24.6 (13.5-48.2) |
| β2-microglobulin, mg/L | 3.37 (2.7-4.7) |
| BM infiltration, % | 55 (25-80) |
| Genomic (NGS), n (%) | |
| MYD88MUT | 48 (97.9) |
| MYD88L265P/MYD88WT | 46 (93.9)/1 (0.02) |
| CXCR4MUT* | 27 (56.2) |
| CXCR4NS/CXCR4FS | 20 (41.6)/7 (14.5) |
| TP53MUT† | 11 (24.4) |
| No. of previous treatments, median | |
| 1 line/2 lines/3 lines | 30/12/6 |
| DRC/other R-chemotherapy/ibrutinib | 19/25/3 |
| Characteristics | Values |
|---|---|
| Age, y | 68.5 (50-83) |
| Males/females, n | 36/13 |
| ECOG PS, 0/1/2/unknown, n | 14/26/8/1 |
| Adenopathy/splenomegaly/hepatomegaly, n | 18/6/3 |
| White blood cells, ×109/L | 5.25 (4.1-7.3) |
| Hemoglobin, g/dL | 10 (8.9-11.1) |
| Platelets, ×109/L | 189 (94-2751) |
| Serum IgM, g/L | 24.6 (13.5-48.2) |
| β2-microglobulin, mg/L | 3.37 (2.7-4.7) |
| BM infiltration, % | 55 (25-80) |
| Genomic (NGS), n (%) | |
| MYD88MUT | 48 (97.9) |
| MYD88L265P/MYD88WT | 46 (93.9)/1 (0.02) |
| CXCR4MUT* | 27 (56.2) |
| CXCR4NS/CXCR4FS | 20 (41.6)/7 (14.5) |
| TP53MUT† | 11 (24.4) |
| No. of previous treatments, median | |
| 1 line/2 lines/3 lines | 30/12/6 |
| DRC/other R-chemotherapy/ibrutinib | 19/25/3 |
Data are median (IQR), unless otherwise noted. Median (IQR) and percentages were calculated for the entire cohort (49 patients), unless otherwise noted. Among the 3 cases of non-MYD88L265P, NGS identified 1 case with the MYD88M232T variant and 1 had a TG→CT substitution at position 38182641, which predicted the MYD88L265P variant.
DRC, dexamethasone-rituximab-cyclophosphamide; R-chemotherapy, rituximab chemotherapy.
Forty-eight patients were evaluated for CXCR4 mutation.
Forty-five patients were evaluated for TP53 mutation.
Treatment
The study flowchart is displayed in Figure 1. Among the 49 enrolled patients, 1 died (unknown cause) before any treatment and 48 patients received the combination induction. Among the 48 treated patients, induction was interrupted in 14 (7 for progressive disease and 7 for AEs). Maintenance was started in 27 patients. At the time of analysis (20 May 2020), 9 patients had completed maintenance and 18 patients had stopped treatment (13 for intolerance and 5 for progression).

Study flowchart, patient disposition, and treatment exposure. M8, month 8; PD, progressive disease.
Study flowchart, patient disposition, and treatment exposure. M8, month 8; PD, progressive disease.
Efficacy
Responses.
Forty-three patients were evaluable after induction for response assessment at month 8, with the 6 nonassessed patients considered failures. The best responses, reached after a median of 6.5 months (IQR, 3.4-7.1; range, 2.6-22.1 months), were very good partial response (VGPR) in 5 patients, partial response (PR) in 27 patients, and MR in 3 patients, leading to ORR and major response rate (MRR) estimates of 71.4% (95% confidence interval [CI], 56.7-83.4) and 65.3% (95% CI, 50.4-78.3), respectively. Stable disease and progressive disease were observed in 5 patients and 3 patients, respectively.
Survival.
With a median follow-up time of 25.9 months (IQR, 22.3-30.5), median PFS was 25.4 months (95% CI, 15.7-29.0) and the PFS rates at 12 and 24 months were 75.5% (95% CI, 64.4-88.6) and 55% (95% CI, 42.7-70.9), respectively (Figure 2A). Median TTF was 25.6 months (95% CI, 19.9-30.4), and the cumulative incidence of new treatment at 12 and 24 months was 22.5% (95% CI, 11.9-35.0) and 36.8% (95% CI, 23.4-50.2), respectively. OS rates at 12 and 24 months were 97.8% (95% CI, 94.1-100) and 89.8% (95% CI, 81.7-98.7), respectively (Figure 2B). Altogether, 28 patients progressed. The outcome of the 3 patients previously exposed to ibrutinib is summarized in supplemental Figure 2. Among the 24 patients who received a new treatment, 14 received a chemotherapy-free regimen (ibrutinib [n = 10], 9 still in response; bortezomib + dexamethasone + rituximab [n = 1]; rituximab [n = 1]; idelalisib [n = 1]; bispecific mAb [n = 1]), and 10 received an immunochemotherapy combination. Of note, IgM rebound was observed after idelalisib discontinuation for toxicity in 4 patients who became symptomatic again and received a new treatment at 2, 8, 12, or 14 months after progression. Two other patients completed maintenance and presented an IgM rebound treated by the reintroduction of idelalisib and ibrutinib. Finally, 6 patients died, 2 free from relapse (1 from unknown cause before protocol therapy was started and 1 from infection after protocol ending), 1 from progression and 3 from infection (n = 2) or from thrombosis (n = 1) after progression.
![OS and PFS according to molecular profile. PFS (A), OS (B), PFS according to CXCR4 mutational status and the type of CXCR4 mutation (frameshift vs non-sense) (C), and PFS according to TP53 mutational status (D). (C) Red line, absence of CXCR4 mutation [CXCR4(WT)] (left panel), green line, CXCR4 mutation non-sense [CXCR4(NS)]; blue line, CXCR4 mutation frameshift [CXCR4(FS)]. (D) Red line, absence of TP53 mutation (WT); blue line, TP53 mutation (MUT).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/9/10.1182_bloodadvances.2020003895/1/m_advancesadv2020003895f2.png?Expires=1765882165&Signature=dl~Y4G6rxAC18QivtXR1PEw~66FA31TN2CcxJ0FXdfjUmREmyvI-JmyFpMjFYxcDv2aTyQa-5kHyglWFSJ658IMxMH--yp1a53qZR9DxLCVKZN7j50rW9UjN63h4xNi6d3cad6eDa0RbqTEJh-On1M1kybkecwgbTUyW6NTelm7zT48LvY-LcUO8OLuN3z5zEndFIIb0drhezEANgCNNcjhOfk6m1RF2L-UcAnx6x1azBu7Y3YzPTZaBddvk8QmBmzcxhJS6J03NjLbaGDBY~6lxpXK6nhxl17qMzDdZWvr8qrLHU~VhIA5xVAvOXzj5b52vwX1B42hx5BLd3zYsNw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
OS and PFS according to molecular profile. PFS (A), OS (B), PFS according to CXCR4 mutational status and the type of CXCR4 mutation (frameshift vs non-sense) (C), and PFS according to TP53 mutational status (D). (C) Red line, absence of CXCR4 mutation [CXCR4(WT)] (left panel), green line, CXCR4 mutation non-sense [CXCR4(NS)]; blue line, CXCR4 mutation frameshift [CXCR4(FS)]. (D) Red line, absence of TP53 mutation (WT); blue line, TP53 mutation (MUT).
OS and PFS according to molecular profile. PFS (A), OS (B), PFS according to CXCR4 mutational status and the type of CXCR4 mutation (frameshift vs non-sense) (C), and PFS according to TP53 mutational status (D). (C) Red line, absence of CXCR4 mutation [CXCR4(WT)] (left panel), green line, CXCR4 mutation non-sense [CXCR4(NS)]; blue line, CXCR4 mutation frameshift [CXCR4(FS)]. (D) Red line, absence of TP53 mutation (WT); blue line, TP53 mutation (MUT).
Impact of molecular profile on response and survival.
Univariate analysis focusing on molecular screening found no significant impact on response rate, depth of response, time to response and PFS of either CXCR4 (whatever the type of mutation) or TP53 genotypes (Figures 2C-D and 3; supplemental Figure 1; supplemental Table 1). The only predictive factor of response was a high level of BM infiltration that was associated with a decreased occurrence of MRR. No prognostic factors for survival were found, except a deleterious effect of TP53 mutation that was nonetheless not statistically significant in the Cox model (supplemental Table 1).
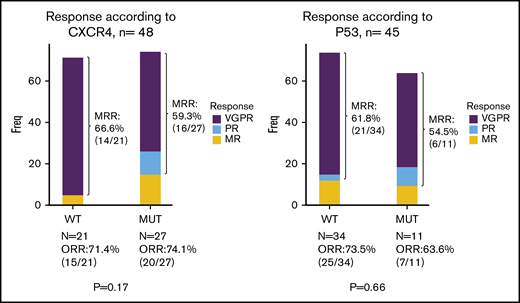
Best responses according to CXCR4 (left panel) and TP53 (right panel) mutational status (n = 49 and n = 45, respectively).
Best responses according to CXCR4 (left panel) and TP53 (right panel) mutational status (n = 49 and n = 45, respectively).
Safety.
After enrollment of the first 25 patients, the DSMB validated the study continuation based on safety data and the safety profile of the combination treatment, without a need for any unplanned additional meetings. Altogether, 124 grade ≥ 3 AEs and 56 SAEs were reported in 43 and 36 patients, respectively. The most prevalent AEs are reported in Table 2. Based on the Bayesian inference, the posterior probability of experiencing ≥1 grade 3-4 AE was 87.0% (95% credibility interval, 76.4-94.7), with a 23% probability that 9 of 10 patients would experience such events. The probability that >3 of 4 patients would experience grade 3-4 AEs was 98%.
Primary AEs observed in patients
| AE | Total AEs (N = 279) | Grade 1-2 (n = 155) (55.5%) | Grade 3-4 (n = 124) (44.5%) | SAEs (n = 56) |
|---|---|---|---|---|
| Hematologic | 82 (29.4) | 24 (8.6) | 58 (20.8) | 2 (3.6) |
| Neutropenia | 54 (19.4) | 9 (3.2) | 45 (16.1) | 1 (1.8) |
| Anemia | 16 (5.7) | 9 (3.2) | 7 (2.5) | 1 (1.8) |
| Thrombocytopenia | 12 (4.3) | 6 (2.2) | 6 (2.2) | 0 |
| Nonhematologic | 197 (70.6) | 131 (46.9) | 66 (23.6) | 54 (96.4) |
| Diarrhea | 24 (8.5) | 9 (3.2) | 15 (5.3) | 12 (21.4) |
| Hepatic cytolysis | 26 (9.3) | 7 (2.5) | 19 (6.8) | 4 (7.1) |
| Infections | 52 (18.6) | 44 (15.7) | 8 (2.8) | 13 (23.2) |
| Skin | 16 (5.7) | 11 (3.9) | 5 (1.8) | 3 (5.3) |
| IRR | 15 (5.4) | 15 (5.3) | 0 | 1 (1.8) |
| Others | 64 (22.9) | 45 (16.4) | 19 (6.8) | 21 (37.5) |
| AE | Total AEs (N = 279) | Grade 1-2 (n = 155) (55.5%) | Grade 3-4 (n = 124) (44.5%) | SAEs (n = 56) |
|---|---|---|---|---|
| Hematologic | 82 (29.4) | 24 (8.6) | 58 (20.8) | 2 (3.6) |
| Neutropenia | 54 (19.4) | 9 (3.2) | 45 (16.1) | 1 (1.8) |
| Anemia | 16 (5.7) | 9 (3.2) | 7 (2.5) | 1 (1.8) |
| Thrombocytopenia | 12 (4.3) | 6 (2.2) | 6 (2.2) | 0 |
| Nonhematologic | 197 (70.6) | 131 (46.9) | 66 (23.6) | 54 (96.4) |
| Diarrhea | 24 (8.5) | 9 (3.2) | 15 (5.3) | 12 (21.4) |
| Hepatic cytolysis | 26 (9.3) | 7 (2.5) | 19 (6.8) | 4 (7.1) |
| Infections | 52 (18.6) | 44 (15.7) | 8 (2.8) | 13 (23.2) |
| Skin | 16 (5.7) | 11 (3.9) | 5 (1.8) | 3 (5.3) |
| IRR | 15 (5.4) | 15 (5.3) | 0 | 1 (1.8) |
| Others | 64 (22.9) | 45 (16.4) | 19 (6.8) | 21 (37.5) |
Only AEs with rates ≥ 5% are reported. Some patients exhibited grade 1-2 and grade 3-4 AEs, so that patient numbers do not add up to the total by row. Eight other malignancies were observed: 5 skin cancer (3 basocellular, 2 epidermoid), 1 prostatic adenocarcinoma, 1 myelodysplastic syndrome, and 1 glioblastoma.
IRR, infusion-related reaction.
No grade 5 toxicity, tumor lysis syndrome, or flare was reported. Four grade 2 CMV reactivations were reported, leading to introduction of an empiric antiviral treatment with a temporary discontinuation of idelalisib. Twenty-six patients (53%) discontinued idelalisib for toxicity after a median of 3 cycles (IQR, 2-4; range, 1-5). Median duration of idelalisib exposure was 8.1 months (IQR, 3.2-15.3). Dose reduction was required in 21 (43%) patients.
Discussion
We report a prospective study of the combination of idelalisib + obinutuzumab in symptomatic R/R WM patients with a 2-year fixed duration of idelalisib. We aimed to demonstrate that median PFS could be improved up to 25 months. This hypothesis was confirmed by our study, with an estimated median PFS of 25.4 months, although likely values given by the 95% CI ranged from 15.7 to 29.0 months. In previously treated WM patients, reuse of a rituximab-based regimen with cyclophosphamide, bendamustine, or proteasome inhibitor was a therapy option if the response lasted > 3 years, whereas ibrutinib alone, especially in rituximab-refractory patients, or in combination with rituximab are also acceptable options.23 Ibrutinib is used in R/R WM patients with a high response rate and a median PFS not reached at 5 years.24 There is actually no consensus in this setting with regard to the duration of treatment (ie, fixed vs until progression).
Idelalisib reversibly inhibits the p110δ-PI3K isoform, leading to decreased phosphorylation of several downstream targets, including Akt, directly inducing apoptosis and ultimately disrupting interactions between tumor cells and the BM microenvironment.13,25 The PI3K/AKT signaling pathways are hyperactivated in WM, highlighting the interest in PI3K inhibitors for WM.26 Considering its combination with obinutuzumab, PI3Kδ inhibition by idelalisib has minimal impact in vitro on the immune effector function of obinutuzumab and rituximab, as measured in natural killer cell–mediated antibody-dependent cellular cytotoxicity, macrophage-mediated antibody-dependent cellular cytotoxicity (ADCP), and whole-blood B-cell depletion.27
Data on efficacy and toxicity in WM patients are scarce, with no data available on the combination with obinutuzumab in this setting. The largest study reported 80% ORR in 10 patients with lymphoplasmacytic lymphoma/WM among 125 low-grade lymphomas that was refractory to anti-CD20 and alkylating agents and treated with idelalisib (150 mg twice a day by mouth).14 These results were updated in 2019 with a median duration of therapy in WM of 29 months (range, 6.4-51). Of the 9 responders, 7 had PR, 1 had MR, and 1 had SD. Median PFS was 22 months, and the median duration of response was 20 months. All patients experienced ≥1 AE related to the treatment, and >65% experienced a grade ≥3 AE. The most common grade ≥3 AEs included neutropenia (28%), diarrhea (28%) and increased liver enzymes (16%). Among patients on therapy for 6 months, AE led to dose reductions for 19.2%, treatment discontinuation for 23.1%, and death for 10.3%.28 A phase 2 study aimed at evaluating idelalisib, 150 mg twice a day by mouth, in R/R WM patients was terminated early because of grade ≥3 increased liver enzyme levels in 3 of the first 5 enrolled patients.16
The pivotal ibrutinib study, which was recently updated,28 and other trials with new BTK inhibitors (monotherapy-like zanubrutinib29 and acalabrutinib30 ) have provided evidence that BTK inhibitors produce long-term disease control as single-agent therapy with manageable safety profiles in treatment-naive or R/R WM patients; most clinicians consider them the treatment of choice in this patient group. Overall, MRR was 80%, with a median follow-up of 27 to 59 months. With regard to BTK inhibitors, the impact of MYD88 and CXCR4 mutations on the quality and duration of the response remains controversial. The impact of MYD88MUT and CXCR4MUT status was reported in patients treated with ibrutinib. Response rates, depth of response, and time to response were highly dependent on the MYD88 and CXCR4 genotype. MYD88WT patients appear to have a lower response rate to ibrutinib.7,11,28 However, the results of the iNNOVATE study combining ibrutinib + rituximab questioned the impact of MYD88MUT status on the response, as well as the second generation of BTK inhibitors, such as acalabrutinib or zanubrutinib, with a high response rate in MYD88WT patients in the ASPEN study.8,10,31 More data are necessary to confirm whether or not MYD88MUT genotype has a negative impact. Of 49 included patients, only 1 had a confirmed MYD88WT genotype, which is insufficient to draw any conclusion for this subgroup.
Ibrutinib-treated CXCR4MUT patients experienced a longer median time to reach MR and lower MRR, including attainment of VGPR, in previously treated WM.11,32,33 Faster IgM response kinetics were also observed for patients with rituximab-refractory CXCR4WT disease who received ibrutinib monotherapy.9 The frequency of CXCR4MUT patients in our trial was higher than previously described (56%), likely as a result of the sensitivity of the deep NGS strategy and digital PCR, as well as the relapse setting.34,35 We did not find any difference in time to response, quality of response, or PFS according to CXCR4 mutation profile or type of mutation (frameshift [FS] vs nonsense [NS]), in contrast to previously described patients treated with ibrutinib.35 TP53 mutations were observed in 24% of the patients in our R/R cohort, which is also higher than previously described at diagnosis.21,36 Sequential TP53 analysis was recently reported in a cohort, including 14 patients with WM, and the acquisition of TP53 variation over time was observed in 29% of cases.37 Selection of the TP53 clone is well described under chemotherapy pressure in other B-cell lymphoproliferative disorders pretreated with genotoxic agents.22,36 The poor prognostic impact of the TP53 mutation has primarily been described in a retrospective analysis of untreated patients or after immunochemotherapy but not in patients treated with BCR signaling inhibitors.21,36 In our study, there was no evidence of any predictive value of TP53 mutation for response or PFS in this small subgroup of patients.
This study has limitations but suggests new treatment opportunities. First, with a median PFS of 25.5 months, we reached our primary goal of demonstrating a median PFS ≥25 months compared with 15 months with usual therapy. Second, the sample size does not allow firm conclusions about the impact of genotype on efficacy, because the study was not designed for this end point. Our results suggest that this combination might be of interest in CXCR4MUT WM patients with advanced disease, because they demonstrate reduced response to ibrutinib. Compensatory PI3K/Akt signaling may contribute to ibrutinib resistance in the CXCR4MUT WM model, as well as in the CXCR4WT WM model, resulting in synthetic lethality toward the PI3K/Akt inhibitor.38-40 Therefore, targeting of the PI3K/Akt pathway could be an option in BTK inhibitor–resistant WM patients and appears to be a proof of concept regarding the efficacy of this class of compound in WM patients. In addition, for the first time, we provide an estimate of the response rate or PFS achieved with a combination regimen including inhibitors of BCR-associated kinases in TP53MUT WM patients; however, the number of patients was small, and a nonstatistically significant deleterious effect of TP53 mutation was observed. Third, the safety of the combination with high liver and gastrointestinal toxicity led to early treatment discontinuation and/or a dose reduction in half of the patients. Using Bayesian analysis, which quantified, through probabilistic statements, the proportion of treated patients who experienced grade 3 or 4 AEs, it appeared more than likely (98%) that more than three fourths of patients could be impacted by such events. These results did not differ substantially from those observed by Gopal et al14 and Wagner-Johnston et al.28 It is a limitation of further clinical use. However, considering the potential efficacy of this class of agents, next-generation PI3K inhibitors associated with obinutuzumab could overcome the toxicity profile observed in this phase 2 trial and may be a reasonable treatment option when standard therapies have been exhausted.
In summary, our study demonstrates interesting efficacy of the combination of idelalisib + obinutuzumab in symptomatic R/R WM patients, although toxicity is a major limitation. Nevertheless, there is a need to develop new approaches in patients who are refractory to BTK inhibitors. This population may benefit from this therapeutic option, which could be investigated in further clinical trials.
Presented in abstract form at the 10th International Workshop on Waldenström’s Macroglobulinemia, New York, NY, 12 October 2018; the 62nd annual meeting of the American Society of Hematology, Orlando, FL, 7 December 2019; and the 40th French Society of Hematology Annual Congress, Paris, France, 11 September 2020.
Data sharing requests should be sent to Cécile Tomowiak (ceciletomowiak@gmail.com).
Acknowledgments
The authors thank all of the French Innovative Leukemia Organization investigators who participated in the REMODEL trial. They also thank Christophe Roumier and the tumor bank of Lille University Hospital (certification NF 96900-2014/65453-1) for handling, conditioning, and storing patient samples. The work of all clinical research assistants is also acknowledged. The authors also thank Jeffrey Arsham, an American medical translator under contract at the Poitiers CHU, for review of our original English-language document.
This work was supported in part by INCA-DGOS-INSERM-12560. SiRIC CURAMUS is financially supported by the French National Cancer Institute, the French Ministry of Solidarity and Health, and INSERM, with financial support from ITMO Cancer AVIESAN (Alliance Nationale pour les Sciences de la Vie et de la Santé/National Alliance for Life Sciences and Health). This study was also supported by Roche and Gilead Sciences, and they provided drugs.
Authorship
Contribution: C.T., S.P., S.C., and V.L. conceived and designed the study, analyzed and interpreted data, and wrote the manuscript, all authors collected and assembled data and approved the final version of the manuscript; and C.T., S.P., D.N., S.C., and V.L. are accountable for all aspects of the work.
Conflict-of-interest disclosure: C.T. has served on Advisory Boards and received travel support from Roche, Gilead Sciences, Janssen, AbbVie, BeiGene, and Takeda. C.H. has received research funding from Takeda and AbbVie and has received honoraria and nonfinancial support from Roche, Janssen-Cilag, Takeda, and AbbVie. P.M. has served on Advisory Boards for BeiGene, Janssen, and Amgen. O.T. has received honoraria and travel support from AbbVie, Takeda, Gilead Sciences, and Janssen and has received honoraria from Sandoz. O.C. has acted as a consultant for and received honoraria from Roche, Gilead Sciences, Bristol Myers Squibb, Takeda, MSD, and AbbVie and has received research funding from Roche, Gilead Sciences, and Takeda. V.L. has acted as a consultant for and received honoraria from Roche, Gilead Sciences, AbbVie, Eli Lilly and Company, Janssen, and AstraZeneca. The remaining authors declare no competing financial interests.
Correspondence: Cécile Tomowiak, Service d'Hématologie et Thérapie Cellulaire, PRC, Hôpital de La Milétrie, Faculté de médecine and INSERM CIC 1402, CHU, 2 Rue de la Milétrie, 86021 Poitiers Cedex, France; e-mail: cecile.tomowiak@chu-poitiers.fr.

