

Key Points
Platelet G protein–coupled receptors regulate the progrowth effect of platelets on ovarian cancer.
The deficiency of G13 or Gi reduces tumor growth and decreases the extravasation of platelets into the tumor.

Abstract
We and other investigators have shown that platelets promote metastasis and the growth of tumors. Our rationale for conducting this study is that platelets’ prometastatic and progrowth effects depend on a close encounter between platelets and cancer cells. This interaction occurs inside blood vessels with circulating tumor cells and outside blood vessels with cancer cells residing in the tumor parenchyma. Our hypothesis was that platelet extravasation is required for the effect of platelets on tumor growth. Platelets respond to environmental stimuli by activation of G protein–coupled receptors on their surface. We investigated the impact of various platelet G proteins on the growth of ovarian cancer tumors and platelet extravasation. We used mice with platelet-specific deficiency of Gαi2 (Gi), Gα13 (G13), or Gαq (Gq) in a syngeneic ovarian cancer model. We measured the total weight of tumor nodules resected from tumor-bearing mice. We developed methods for automated whole-slide image acquisition and unbiased computerized image analysis to quantify extravasated platelets. We compared the number of platelets inside tumor nodules of platelet G protein–deficient tumor-bearing mice. We found that deficiency of Gi and G13, but not Gq, in platelets resulted in smaller tumors compared with those in corresponding littermates. Deficiency of Gi and G13 in platelets reduced the number of extravasated platelets by >90%, but deficiency of Gq did not reduce the number of extravasated platelets significantly. The lack of Gi or G13 in platelets reduced platelet extravasation into the tumor and tumor growth.
Introduction
Many cancer patients develop elevated platelet counts (thrombocytosis) that are associated with a worse prognosis.1 The role of platelets in cancer and the interactions between cancer cells and platelets have been investigated for a long time.1-3 The effect of platelets on cancer progression was primarily attributed to promoting metastasis via direct interaction with circulating cancer cells4,5 and endothelial cells.6 However, we have shown that platelets also increase the growth of ovarian cancer.7,8 Other investigators have shown a similar effect in different cancer types.9-11 For platelets to directly affect the primary tumors, they should be present in the tumor microenvironment. The presence of extravascular platelets in the parenchyma of tumor specimens from tumor-bearing mice and patients has been reported by our group and by other investigators.12-15 We speculated that platelets interact with circulating tumor cells and, after extravasation, with the cancer cells in the primary tumor.
In this study, we examined whether the progrowth effect of platelets on ovarian cancer and the number of platelets extravasated into tumors depend on platelet G proteins.
We have shown that adenosine diphosphate (ADP) secreted from ovarian cancer cells can activate platelets.8 ADP can bind to P2Y12 and P2Y1 on platelets; however, the impact of ADP secreted from cancer cells on the platelet-mediated protumor effect was dependent on P2Y12. The absence or inhibition of P2Y12 reduced the growth-promoting effect that platelets had on ovarian cancer. On the other hand, deficiency of P2Y1 did not have any effect on tumor growth. Although P2Y12 and P2Y1 are G protein–coupled receptors (GPCRs) and bind to ADP, their postreceptor events are distinct. P2Y12 signals through Gi protein, and P2Y1 signals through Gq.
Platelets become activated in response to different agonists, such as thrombin, ADP, arachidonic acid, epinephrine, and collagen, through GPCRs. Thrombin activates platelets via protease-activated receptor 1 (PAR-1) and PAR-4 in humans (PAR-3 and PAR-4 in mice), ADP activates platelets through P2Y12 and P2Y1, arachidonic acid activates platelets via thromboxane A2 receptor, and epinephrine activates platelets via α2A adrenergic receptor. G proteins are heterotrimeric protein complexes that are composed of α, β, and γ subunits. Upon activation of a GPCR, its Gα subunit separates from the β and γ subunits and initiates intracellular signaling. GPCRs are distinguished by their Gα subunits. There are 3 groups of Gα proteins: Gαi, Gαq, and Gα13. In platelets, the most common Gαi is Gαi2, which couples to P2Y12. Gαq couples to PARs, thromboxane A2 receptor, and P2Y1, whereas Gα13 couples to PARs. Because one ligand can bind to different GPCRs and one GPCR can bind to different G proteins, we hypothesized that, ultimately, G proteins determine the effect of a GPCR ligand on tumor growth. We investigated the role of various G proteins in platelets in the progression of cancer and on the extravasation of platelets, using mice with platelet-specific Gαi2 deficiency (Gnαi2fl/fl; PF4-cre+ mice), Gαq deficiency (Gαqfl/fl; PF4-cre+), or Gα13 deficiency (Gα13fl/fl; PF4-cre+)16,17 in murine models of ovarian cancer. For simplification, Gαi2, Gαq, and Gα13 are hereafter referred to as Gi, Gq, and G13, respectively.
Materials and methods
All mice studies were conducted according to the protocols approved by the Institutional Review Board and the Institutional Animal Care and Use Committee of the University of Texas MD Anderson Cancer Center.
Cell lines and reagents
ID8 murine ovarian cancer cells were maintained in Dulbecco modified Eagle medium supplemented with 10% heat-inactivated fetal bovine serum, 1% penicillin-streptomycin, and 0.1% insulin-transferrin–sodium selenite at 37°C in a humidified incubator infused with 20% O2 and 5% CO2.
Murine model of ovarian cancer
Two million ID8 murine ovarian cancer cells were resuspended in 200 µL of Hanks balanced salt solution and injected into the peritoneal cavity of 6- to 8-week-old female mice, as described previously.8 The platelet-specific Gαi2 protein–knockout, Gnαi2fl/fl; PF4-cre+ mice (Gαi2−/− or simply Gi−/−) on a C57BL/6 genetic background16 and frozen sperm from Gα13fl/fl; Gαq fl/fl; PF4-cre+ mice (G13−/−; Gq−/−)17 were from B.N. and Stefan Offermanns (Max Planck Institute for Heart and Lung Research, Bad Nauheim, Germany), respectively. Rederivation of G13−/− (G13fl/fl; PF4-cre+) mice and Gq−/− (Gqfl/fl; PF4-cre+) mice was performed by the Genetically Engineered Mouse core-Facility at the University of Texas MD Anderson Cancer Center. Gα13fl/fl; Gαqfl/fl; PF4-cre+ (G13−/−, Gq−/−) double knockout mice were separated to a single gene knockout (G13−/− or Gq−/−) mice. After rederivation, newborn mice were genotyped according to previously published protocols.16,17 Littermates of mice without PF4-cre (Gnαi2fl/fl and Gα13fl/fl; Gαqfl/fl) were used as controls. Wild-type C57BL/6 mice were purchased from the Jackson Laboratory for the rederivation procedure.
After injection, mice were monitored weekly for 4 weeks and then daily for another 1 to 2 weeks until they became moribund. We euthanized moribund mice; tumor nodules were resected, weighed, and fixed in 10% formalin for histological analysis.
Immunofluorescence staining
Immunofluorescence staining for CD31 and CD42b was performed on 4-µm-thick sections of formalin-fixed paraffin-embedded tumor nodules using a previously described method.8 Briefly, deparaffinized tumor slides underwent antigen retrieval. After blocking nonspecific binding, slides were incubated with primary CD31 (RB-10333; 1:50 dilution; Thermo Fisher Scientific, Waltham, MA) antibody overnight at 4°C. The next day, slides were washed and incubated with the appropriate fluorophore secondary Alexa Fluor 647–conjugated antibody for 1 hour. Then, the slides were washed and incubated with FITC-conjugated anti-CD42b (antibody X488; 1:25 dilution; emfret Analytics, Eibelstadt, Germany) overnight. After another washing step, slides were mounted in a soluble antifade mounting buffer. Acquired images were analyzed to quantify extravasated platelets, as described in supplemental Methods and supplemental Figures.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 8 software. A 2-tailed Student t test or Welch's t test was used to determine the statistical significance of comparisons. For all statistical analyses, P < .05 was considered significant.
Results and discussion
Various platelet agonists (ADP, thrombin, and thromboxane A2) activate platelets by binding to GPCRs on the surface of platelets, which results in shape change, degranulation, and aggregation.18 We examined the effect of platelet-specific G proteins on tumor growth in murine models of ovarian cancer. ID8 murine ovarian cancer cells were injected into the peritoneal cavity of platelet-specific G protein–knockout mice: Gi−/−, G13−/−, or Gq−/−. Deficiency of Gi, G13, or Gq in platelets did not change their number in the peripheral blood. Platelet-specific Gi−/− mice (Gi△/△) and G13−/− mice (G13△/△) developed smaller tumors compared with control littermates (Gifl/fl and G13fl/fl, respectively). However, platelet-specific Gq−/− (Gq△/△) tumor-bearing mice and control littermates (Gqfl/fl) developed tumors that were the same size (Figure 1A). The proliferation rate of cancer cells inside tumor nodules, quantified by the percentage of Ki67 positivity (supplemental Methods), was significantly reduced in Gi△/△ and G13△/△ mice, but it was not changed in Gq△/△ tumor-bearing mice, compared with the corresponding controls (Figure 1B). We measured the effect of G protein–deficient platelets on the induction of an invasive phenotype in ID8 cells in vitro using a Transwell migration assay (supplemental Methods). ID8 cells migrate less after incubation with Gi- or G13-deficient platelets compared with control platelets. Lack of Gq in platelets did not affect the migration of coincubated cancer cells (Figure 1C).
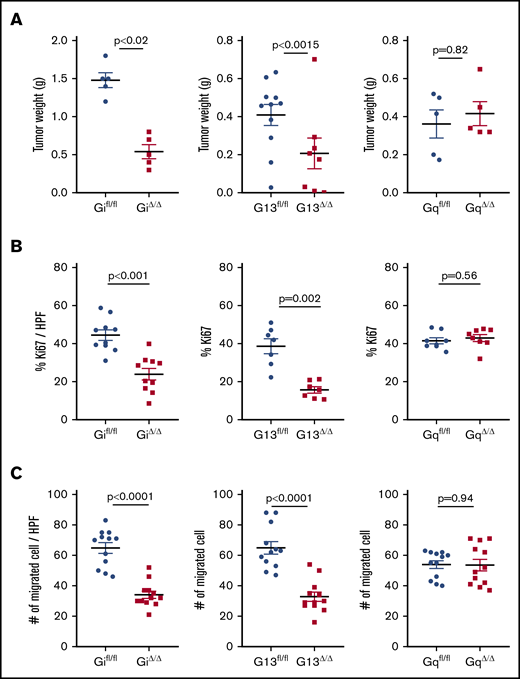
Platelet G proteins and growth of orthotopic ovarian cancer tumors in mice and viability of cancer cells. (A) Gi△/△ mice (left panel) and G13△/△ mice (middle panel) developed smaller tumors 6 to 8 weeks after intraperitoneal injection of ID8 murine ovarian cancer cells (Gi△/△, 0.54 ± 0.1 g; Gifl/fl, 1.46 ± 0.1 g; G13△/△, 0.17 ± 0.007 g; G13fl/fl, 0.42 ± 0.065 g). The deficiency of Gq protein in platelets (Gq△/△) did not affect the final tumor size in mice (Gq△/△, 0.41 ± 0.07 g; Gqfl/fl, 0.48 ± 0.11 g) (right panel). n = 6 to 8 mice per group. (B) The proliferation rate of cancer cells inside tumor nodules was quantified by the percentage of Ki67 positivity per high-power field (HPF): Gifl/fl, 45 ± 4%; Gi△/△, 24 ± 5%; G13fl/fl, 39 ± 5%; G13△/△, 16 ± 2%; Gqfl/fl, 42 ± 2%; Gq△/△, 43 ± 3%). Three tumor-bearing mice, 1 nodule from the mouse, and 3 or 4 HPFs per nodule were examined. (C) Invasiveness of cancer cells after incubation with G protein–deficient platelets was measured in vitro by migration assay: Gifl/fl: 65 ± 6 per HPF; Gi△/△, 34 ± 4 per HPF; G13fl/fl, 65 ± 7 per HPF; G13△/△, 33 ± 5 per HPF; Gqfl/fl, 54 ± 4 per HPF; Gq△/△, 54 ± 7 per HPF. Four HPFs from 3 membranes were imaged and analyzed. All data are mean ± standard deviation. A 2-tailed Student t test was used to determine statistical significance; P < .05 was considered significant.
Platelet G proteins and growth of orthotopic ovarian cancer tumors in mice and viability of cancer cells. (A) Gi△/△ mice (left panel) and G13△/△ mice (middle panel) developed smaller tumors 6 to 8 weeks after intraperitoneal injection of ID8 murine ovarian cancer cells (Gi△/△, 0.54 ± 0.1 g; Gifl/fl, 1.46 ± 0.1 g; G13△/△, 0.17 ± 0.007 g; G13fl/fl, 0.42 ± 0.065 g). The deficiency of Gq protein in platelets (Gq△/△) did not affect the final tumor size in mice (Gq△/△, 0.41 ± 0.07 g; Gqfl/fl, 0.48 ± 0.11 g) (right panel). n = 6 to 8 mice per group. (B) The proliferation rate of cancer cells inside tumor nodules was quantified by the percentage of Ki67 positivity per high-power field (HPF): Gifl/fl, 45 ± 4%; Gi△/△, 24 ± 5%; G13fl/fl, 39 ± 5%; G13△/△, 16 ± 2%; Gqfl/fl, 42 ± 2%; Gq△/△, 43 ± 3%). Three tumor-bearing mice, 1 nodule from the mouse, and 3 or 4 HPFs per nodule were examined. (C) Invasiveness of cancer cells after incubation with G protein–deficient platelets was measured in vitro by migration assay: Gifl/fl: 65 ± 6 per HPF; Gi△/△, 34 ± 4 per HPF; G13fl/fl, 65 ± 7 per HPF; G13△/△, 33 ± 5 per HPF; Gqfl/fl, 54 ± 4 per HPF; Gq△/△, 54 ± 7 per HPF. Four HPFs from 3 membranes were imaged and analyzed. All data are mean ± standard deviation. A 2-tailed Student t test was used to determine statistical significance; P < .05 was considered significant.
Gq-deficient platelets had a significant reduction in degranulation in response to all agonists but retained their ability to change shape in response to thromboxane A2, collagen, and thrombin.19 On the other hand, the impact of Gi and G13 on degranulation was agonist specific. Although Gi deficiency reduced platelet degranulation due to ADP and collagen-related peptide,16 G13 deficiency reduced epinephrine-, U46619-, and thrombin-induced degranulation.20 Overall, although the degranulation of activated platelets depends, to a great extent, on Gq, shape change is primarily dependent on G13. Because G proteins affect activation and shape change in platelets, we examined whether they also affect platelet extravasation into tumors in our murine models.
We have shown a reduction in the number of extravasated platelets in tumor-bearing mice with abnormal platelet function.8,13 We hypothesized that the activation of platelets by environmental stimuli regulates platelet extravasation. We compared extravasation of Gα protein–deficient and wild-type platelets into tumor parenchyma. To reliably and efficiently quantify up to tens of thousands of platelets in a whole-tumor tissue slide is extremely challenging. We developed automated whole-slide imaging and image analysis based on artificial intelligence learning to quantify the number of extravascular platelets in ovarian cancer tissue sections (supplemental Figures 1-3). We measured the density of extravasated platelets in tumor specimens resected from tumor-bearing mice with platelet-specific Gα protein deficiency. Immunofluorescence staining was performed on tumor nodules resected from Gi−/−, G13−/−, or Gq−/− mice, as well as their control tumor-bearing littermates, using an anti-CD42b antibody as a platelet marker and anti-CD31 antibody as an endothelial cell marker. We examined the entire section of a tumor nodule, avoiding any selection bias in choosing the microscopic fields for counting platelets. We found that the lack of Gi and G13 reduced the number of extravascular platelets in the tumor parenchyma (Figure 2). Although platelet density in the tumors resected from Gifl/fl littermates (controls) was 6.12 × 104 per mm2, it was substantially lower in Gi−/− tumor-bearing mice (0.59 × 104 per mm2). There also was a significant reduction in the density of extravasated platelet in tumors from G13−/− mice (0.60 × 104 per mm2) compared with their G13fl/fl littermates (6.5 × 104 per mm2). However, there was not a significant reduction in the number of extravasated platelets in tumors from Gq−/− mice (2.42 × 104 per mm2) compared with their Gqfl/fl tumor-bearing littermates (3.8 × 104 per mm2).
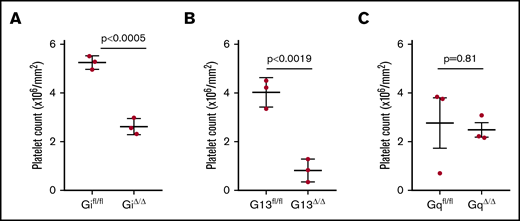
Effect of G protein on platelet extravasation. The number of extravasated platelets (inside tumor parenchymal and outside blood vessels) into tumor nodules of tumor-bearing mice was determined by automatic whole-slide imaging and analysis. The effect of G protein deficiency in platelets was determined by comparing extravasated platelets in mice with platelet-specific Gi deficiency (Gi△/△) (A), G13 deficiency (G13△/△) (B), or Gq deficiency (Gq△/△) (C) with their corresponding littermates. One whole section was imaged and analyzed from each tumor nodule (1 tumor nodule from 3 mice per group). Data are mean ± standard deviation. A 2-tailed Student t test was used to determine statistical significance; P < .05 was considered significant.
Effect of G protein on platelet extravasation. The number of extravasated platelets (inside tumor parenchymal and outside blood vessels) into tumor nodules of tumor-bearing mice was determined by automatic whole-slide imaging and analysis. The effect of G protein deficiency in platelets was determined by comparing extravasated platelets in mice with platelet-specific Gi deficiency (Gi△/△) (A), G13 deficiency (G13△/△) (B), or Gq deficiency (Gq△/△) (C) with their corresponding littermates. One whole section was imaged and analyzed from each tumor nodule (1 tumor nodule from 3 mice per group). Data are mean ± standard deviation. A 2-tailed Student t test was used to determine statistical significance; P < .05 was considered significant.
In our syngeneic murine model of ovarian cancer based on ID8 cells, Gi and G13 in platelets determined extravasation of platelets into tumor and the protumor effect of platelets. However, our conclusion cannot be generalized for all cancer cells lines.15 In different cancer models, platelets may extravasate using other mechanisms, which may have a different effect on the tumor. We propose that the number of extravasated platelets (tumor-infiltrating platelets) may have prognostic value. We put forward the hypothesis that measurement of tumor-infiltrating platelets in the initial biopsy or resected cancer specimens can be a clinically relevant biomarker for predicting the aggressiveness of cancer. This hypothesis will be investigated further by examining specimens from ovarian cancer patients, using the automated quantitative methods presented in this manuscript.
Data sharing requests should be sent to Vahid Afshar-Kharghan (vakharghan@mdanderson.org).
Acknowledgments
The authors thank Stefan Offermanns for generously providing frozen sperm from Gα13fl/fl; Gαqfl/fl; PF4-cre+ mice.
This work was supported in part by the National Cancer Institute of the National Institutes of Health (grants CA177909, CA016672, P50 CA217685, and P50 CA098258 [A.K.S.]; grants CA231141 and CA177909 [V.A.-K.]; and P30 CA016672 [The University of Texas MD Anderson Cancer Center Core grant]), the American Cancer Society (A.K.S.), the Frank McGraw Memorial Chair in Cancer Research (A.K.S.), the American Society of Hematology’s Bridge Fund (V.A.-K.), the Ting Tsung and Wei Fong Chao Foundation (S.T.C.W.), the John S. Dunn Foundation (S.T.C.W.), the Johnsson Estate Fund (S.T.C.W.), and the Advanced Cellular and Tissue Microscopy Core through Houston Methodist Hospital (S.T.C.W.). B.N. was supported by the Deutsche Forschungsgemeinschaft (NU 53/13-1). M.S.C. was supported by the Liz Tilberis Early Career Development Award from the Ovarian Cancer Research Alliance and an Ovarian Cancer Research Grant in honor of Liza Chance from the Foundation for Women’s Cancer. J.L. was supported by a Fellowship from the Cancer Prevention and Research Institute of Texas Computational Cancer Biology Training Program.
Authorship
Contribution: M.S.C. performed research, analyzed data, and wrote the manuscript; J.L. designed research, performed research, analyzed data, and wrote the manuscript; R.G.-D., H.L., T.H., Y.H., K.L., and T.S. performed research; M.V. performed research and analyzed data; B.N. contributed vital new reagents; A.K.S analyzed data and wrote the manuscript; and S.T.C.W. and V.A.-K. designed research, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Vahid Afshar-Kharghan, Section of Benign Hematology, MD Anderson Cancer Center, Z9.5044, 6565 MD Anderson Blvd, Houston, TX 77030; e-mail: vakharghan@mdanderson.org; and Stephen T. C. Wong, Systems Medicine and Bioengineering, Houston Methodist Cancer Center, 6670 Bertner Ave, Room 6.211, Houston, TX 77030; e-mail: stwong@houstonmethodist.org.

