

Key Points
Leishmania infection–induced thrombocytopenia is a consequence of defective platelet production and enhanced platelet clearance.
Thrombocytopenia in visceral leishmaniasis is reversible after the clearance of parasites and restoration of tissue microarchitecture.

Abstract
Visceral leishmaniasis is an important yet neglected parasitic disease caused by infection with Leishmania donovani or L infantum. Disease manifestations include fever, weight loss, hepatosplenomegaly, immune dysregulation, and extensive hematological complications. Thrombocytopenia is a dominant hematological feature seen in both humans and experimental models, but the mechanisms behind this infection-driven thrombocytopenia remain poorly understood. Using a murine model of experimental visceral leishmaniasis (EVL), we demonstrated a progressive decrease in platelets from day 14 after infection, culminating in severe thrombocytopenia by day 28. Plasma thrombopoietin (TPO) levels were reduced in infected mice, at least in part because of the alterations in the liver microenvironment associated with granulomatous inflammation. Bone marrow (BM) megakaryocyte cytoplasmic maturation was significantly reduced. In addition to a production deficit, we identified significant increases in platelet clearance. L donovani–infected splenectomized mice were protected from thrombocytopenia compared with sham operated infected mice and had a greater response to exogenous TPO. Furthermore, infection led to higher levels of platelet opsonization and desialylation, both associated with platelet clearance in spleen and liver, respectively. Critically, these changes could be reversed rapidly by drug treatment to reduce parasite load or by administration of TPO agonists. In summary, our findings demonstrate that the mechanisms underpinning thrombocytopenia in EVL are multifactorial and reversible, with no obvious residual damage to the BM microenvironment.
Introduction
Visceral leishmaniasis (VL) is a neglected parasitic disease caused by the protozoan parasites Leishmania donovani and L infantum. Approximately 50 000 to 90 000 cases and ∼20 000 deaths occur every year, mostly in Brazil, East Africa, and India.1,2 Fever, weight loss, and hepatosplenomegaly are common clinical findings, along with hematological complications and immune dysregulation.3,4 Leishmania reside and replicate inside tissue macrophages as nonmotile amastigote forms and survive by hijacking a variety of innate and acquired immune response pathways at the cellular and tissue level to subvert macrophage leishmanicidal activity.4,5
Thrombocytopenia is one of the predominant hematological features of many viral, bacterial, and parasitic infections with complex underlying pathogeneses.6-8 Infection has been associated with bone marrow (BM) suppression that leads to reduced platelet production, defective thrombopoietin (TPO) production, and enhanced peripheral clearance.8-11 In addition to alterations in platelet production, the evidence supports alterations in the pathways for removal of platelets as an important contributor to thrombocytopenia. For example, antiplatelet antibodies detected in infections such as hepatitis-C,12 HIV,13 and immune thrombocytopenia (ITP) secondary to infection,14 can lead to excessive platelet clearance by tissue macrophages.15 Furthermore, exposure of galactose residues on the membranes of desialylated platelets in sepsis has been shown to make them more prone to clearance by hepatocytes or Kupffer cells, providing an alternate route for platelet clearance outside of the spleen.16-18 Collectively, these changes may lead to disease-related cytopenias and hematological complications.19-22
Thrombocytopenia, along with anemia, is a common clinical finding in patients with VL and in experimental models of disease, with the mechanisms underlying anemia being the more well studied. For example, experimental studies using murine models have shown that L donovani infection triggers emergency hematopoiesis and affects the hematopoietic stem cell (HSC) compartment through exhaustion and reduced potential for self-renewal.23,24 In addition, dysfunctional medullary erythropoiesis may contribute to anemia.25 Little is known, however, about the mechanisms responsible for thrombocytopenia during this disease, beyond the identification of a high prevalence of platelet-binding antibodies in dogs with acute canine VL.26,27 VL is notable in affecting primarily 3 major systemic organs (the liver, BM, and spleen), each of which is also involved in the production, regulation, and clearance of platelets under physiological and pathological conditions. At homeostasis, normal microarchitectural organization of these tissues is critical for the production and clearance of platelets.28-30 During VL, major microarchitectural changes occur, including prolific hepatic granulomatous inflammation,31 BM hypocellularity or myelodysplasia,3,32,33 and remodeling of splenic red and white pulp.34 The impact of these changes in terms of drivers of platelet production and clearance are unknown.35 In particular, splenic macrophages are highly effective at removing aged or damaged blood cells, and these functions can be enhanced as a consequence of infection-associated splenomegaly and bystander cytokine-dependent activation.31,36,37
In this study, we showed that thrombocytopenia is a feature of experimental VL (EVL) in a C57BL/6 mouse model and characterized contributing pathways at the tissue, cellular, and molecular levels. Using a combination of surgical intervention, drug treatment, and cytokine reconstitution, we found that L donovani infection led to alterations in the megakaryocyte (MK) demarcation membrane system (DMS), commensurate with decreased hepatic production and plasma concentrations of TPO and platelet membrane changes that would facilitate enhanced platelet clearance in both spleen and liver. Hence, multiple pathways contribute to the evolution of reversible thrombocytopenia during this infection.
Methods
Ethics statement
Ethical approval for the study was obtained from the Animal Welfare and Ethical Review Board of the Department of Biology, University of York. All procedures were performed under the authority of United Kingdom Home Office Project License P49487014.
Mice, infection, and experimental manipulation
Female C57BL/6 wild-type (WT) mice 6 to 8 weeks of age, bred and maintained at the Biological Services Facility, University of York, were used for all experiments. The mice were housed in individually ventilated cages at 20 to 21°C and 56% humidity under specific-pathogen–free conditions with quarterly health screening (Federation of European Laboratory Animal Science Associations standards 67M and 51M) and provided with food and water ad libitum. Sham operated and splenectomized C57BL/6 mice were purchased from Jackson Laboratories and allowed to recover for 3 weeks before infection. The mice were infected via the IV route with 3 × 107 amastigotes of an Ethiopian strain of L donovani (LV9) without anesthesia. Control age-matched mice were housed in similar conditions. As required, infected mice were treated with a single IV dose (8 mg/kg) of AmBisome (Gilead Sciences International Ltd, Cambridge, United Kingdom), resuspended in sterile 5% dextrose in distilled water. As required, sham operated and splenectomized-infected mice were treated with exogenous human recombinant TPO (rTPO; Zymogenetics, Seattle, WA) at a dose of 50 µg/g bodyweight intraperitoneally, daily for 5 consecutive days. ITP was induced by a single intraperitoneal dose of 0.2 µg/g of rat anti-mouse CD41 monoclonal antibody (MWReg30; BD Pharmingen).38 All mice were euthanized by CO2 inhalation and cervical dislocation at specified times. The parasite burden was determined in Giemsa-stained tissue impression smears and expressed as Leishman-Donovan units (LDUs), using the following formula: LDU = number of amastigotes/1000 cell nuclei × organ weight (grams).
Blood collection and analysis
Blood was collected in EDTA-coated tubes (Microvette CB300 EDTA; Sarstedt, Nümbrecht, Germany) from a lateral tail vein of infected and uninfected mice at specific time points. Blood analysis was performed on a scil Vet abc Plus+ blood counter (Scil Animal Care Company, Dumfries, United Kingdom). For blood smears, air-dried, methanol-fixed blood smear slides were stained with the Hemacolor staining kit (Merck, Darmstadt, Germany) per the manufacturer’s guidelines.
Platelet assays
Mice were bled via cardiac puncture after they were anesthetized by isoflurane inhalation in a secure chamber (Apollo TEC3 Isoflurane Vaporizer; Sound Veterinary Equipment, Rowville, VIC, Australia), and platelets were isolated by using established protocols.39 Freshly isolated platelets were stained with anti-CD41 FITC antibody (1:200; cat no. 133904; BioLegend) for 30 minutes at room temperature (RT). The platelets were incubated with either (30 minutes at RT) conjugated anti-mouse IgG AF647 (1:500; cat no. A-32728; Invitrogen) and IgM PE-Cy7 (1:500; cat no. 552867; BD Biosciences, San Jose, CA) antibodies for anti-platelet antibody detection or biotinylated lectins (Ricinus communis agglutinin-1 [RCA-1] and Maackia amurensis lectin-2 [MAL-2]; 0.5 μg/mL; Vector Laboratories) followed by streptavidin-labeled phycoerythrin (1:200; cat no. 554061; BD Biosciences) for 15 minutes for identifying desialylated platelets. The platelets were washed with buffer (150 nM NaCl and 50 nM HEPES, [pH 6.5] at RT) and acquired on a BD LSR II Fortessa flow cytometer (BD Biosciences). Unstained, single antibody–stained, and neuraminidase-treated (3 U/mL at 37°C for 30 minutes; Invitrogen) platelets were used as controls. Data were analyzed with FlowJo, v10.6.2 software (FlowJo, Ashland, OR) and FCS Express 7 (De Novo Software, Pasadena, CA).
Quantification of circulating TPO
Circulating TPO levels were measured in the serum samples of infected and uninfected mice with a Mouse Thrombopoietin Quantikine ELISA Kit (MTP100; R&D Systems, Minneapolis. MN), per the manufacturer’s guidelines.
Measurement of Thpo mRNA accumulation
RNA was extracted from liver samples with Micro RNeasy kits (Qiagen), per the manufacturer’s guidelines, and RNA quality and quantity were assessed with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). The synthesis of cDNA was performed with a High-Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific) using the manufacturer’s protocol. Duplex quantitative polymerase chain reaction (qPCR) was performed with TaqMan gene expression assays (Applied Biosystems, Thermo Fisher Scientific) with a Thpo FAM and a Hrpt VIC dye-labeled probes. Real-time PCRs (RT-PCRs) were performed on QuantStudio 3 Real-Time PCR system (Applied Biosystems, Thermo Fisher Scientific) under the following cycling conditions: 95°C for 20 seconds followed by 40 cycles of 95°C for 1 second and 60°C for 20 seconds. Data were analyzed using the QuantStudio Design and Analysis software (Thermo Fisher Scientific). Thpo mRNA expression levels were normalized to the Hprt housekeeping gene by using ΔΔCt calculations. Relative expression levels between control and experimental groups were calculated using 2−ΔΔCt.
Histological analysis
The methodology for processing BM and liver samples for light and electron microscopy, and the methodology for image analysis to identify changes in the MK DMS and for evaluating expression of TPO by hepatocytes are provided in the supplemental Methods.
Statistical analysis
All the downstream tissue analyses were performed by researchers blinded to the treatment groups and were analyzed using Prism 8.0 software (GraphPad, San Diego, CA). When comparing 2 groups, the unpaired Student t test or Mann-Whitney U test was applied, depending on the distribution of data. An analysis of variance (ANOVA) was used for multiple comparisons followed by the post hoc Tukey’s test. P < 0.05 was taken as significant.
Results
L donovani infection results in progressive thrombocytopenia
We used a well-characterized C57BL/6 mouse model of L donovani infection to study the mechanisms of thrombocytopenia (Figure 1A). As the early kinetics of infection have been well-described in both spleen and liver,23,25 we performed sequential blood sampling over a 28-day period before termination of the experiment. As expected, at the study end point, high parasite burdens in liver and spleen were recorded, along with significant hepatosplenomegaly (Figure 1B-C) and mild anemia25 (supplemental Figure 1). To evaluate the kinetics of development of thrombocytopenia, blood samples were taken weekly. Platelet counts in uninfected mice remained consistent over the time course of these experiments, indicating that sequential sampling had minimal effects. In infected mice, the platelet count was similar to that of control mice at day 7 post infection (PI). but showed a progressive decline from day 14 PI, with all mice being severely thrombocytopenic by day 28 PI (Figure 1D, left). Platelets in infected mice were also significantly larger than those in uninfected mice (mean platelet volume [MPV]; 7.58 ± 0.36 µm3 vs 5.6 ± 0.2 µm3 in infected vs uninfected mice at day 28 PI; Figure 1D, right). Giemsa-stained blood smears confirmed the presence of giant platelets in infected mice (Figure 1E). In contrast to the findings in C57BL/6 mice, we found that BALB/c mice, another strain susceptible to L donovani infection, showed only minimal levels of thrombocytopenia (supplemental Figure 2). Thus, infection of C57BL/6 mice provides a more appropriate model for the study of this aspect of human disease, in addition to its value in studying the alterations in erythropoiesis that lead to anemia.25
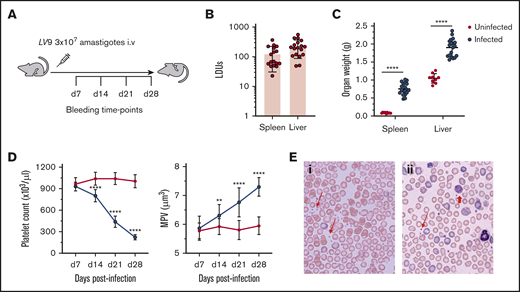
Thrombocytopenia associated with experimental VL. (A) C57BL/6 WT mice were infected with 3 × 107L donovani amastigotes IV for 28 days. Mice were bled every week via a tail vein for platelet counts, and tissues were harvested at day 28 PI for further analysis. (B) The parasite burden was calculated as LDUs on Giemsa-stained liver (n = 18) and spleen (n = 17) impression smears on day 28 PI. (C) Postmortem liver and spleen weights were measured in infected (n = 19) and uninfected (n = 9) mice. (D) Platelet count (left) and MPV (right) were monitored weekly after infection in infected (n = 12) and uninfected (n = 15) mice. (E) Blood smears of uninfected (i) and infected (ii) mice show the presence of giant platelets on the infected mouse blood smear. Representative normal platelets (thin red arrows) and giant platelets (thick red arrow) are indicated. Original magnification ×63. All data were pooled from 3 different independent experiments and analyzed using the unpaired Student t test comparing the mean ± SD of uninfected vs infected at each time point. **P < .01; ****P < .0001.
Thrombocytopenia associated with experimental VL. (A) C57BL/6 WT mice were infected with 3 × 107L donovani amastigotes IV for 28 days. Mice were bled every week via a tail vein for platelet counts, and tissues were harvested at day 28 PI for further analysis. (B) The parasite burden was calculated as LDUs on Giemsa-stained liver (n = 18) and spleen (n = 17) impression smears on day 28 PI. (C) Postmortem liver and spleen weights were measured in infected (n = 19) and uninfected (n = 9) mice. (D) Platelet count (left) and MPV (right) were monitored weekly after infection in infected (n = 12) and uninfected (n = 15) mice. (E) Blood smears of uninfected (i) and infected (ii) mice show the presence of giant platelets on the infected mouse blood smear. Representative normal platelets (thin red arrows) and giant platelets (thick red arrow) are indicated. Original magnification ×63. All data were pooled from 3 different independent experiments and analyzed using the unpaired Student t test comparing the mean ± SD of uninfected vs infected at each time point. **P < .01; ****P < .0001.
BM MK ultrastructure and functional capacity in EVL
We first investigated whether thrombocytopenia is caused by defective production of platelets in mice with L donovani infection by examining the gross and ultrastructural morphology of BM MKs. No significant difference in the number of BM MKs was observed on hematoxylin and eosin–stained BM sections at the time of maximal thrombocytopenia (Figure 2A). Maturation of MKs is characterized by the development of a unique membrane system, the DMS, which ultimately divides the cytoplasm during platelet formation.40 To assess whether the DMS of BM MKs in infected mice was altered, we applied quantitative image analysis to transmission electron microscopy (TEM) images of BM MKs (supplemental Figure 3). Analysis of the perimeter of the DMS indicated that these membranes were significantly reduced in MKs in infected mice, compared with uninfected mice (Figure 2B). Reduction in the DMS of MKs in infected mice suggests that their capacity to produce platelets may be impaired as a consequence of infection. Platelet territories were also less pronounced in MKs in infected mice compared with those in uninfected mice, further suggesting a reduced capacity to produce platelets (Figure 2C).
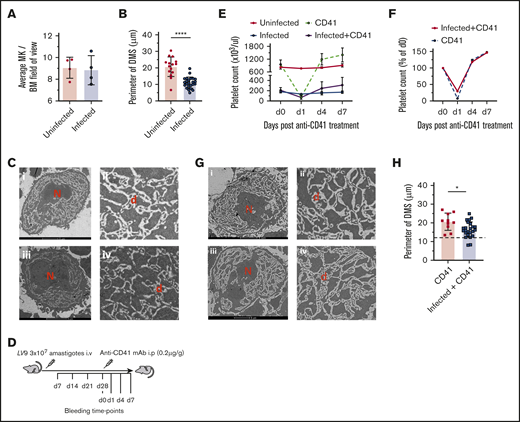
Alteration to BM megakaryocyte form and function during experimental VL. (A) BM MKs were counted manually on hematoxylin and eosin–stained frozen sections in 10 random fields of view for uninfected (n = 4) and infected (n = 4) mice. TEM (FEI Tecnai 12G2) was performed on femur sections of uninfected and day-28–infected mice. Images were analyzed by creating outlines of demarcation membranes with Fiji ImageJ software (supplemental Figure 3). (B) Perimeters were then calculated on 25 uninfected and 46 infected mouse BM MKs. Data represent the mean ± standard error of the mean (SEM) across individual mice. The unpaired Student t test was applied to the means of the perimeter of membranes; ****P < .0001, in uninfected vs infected mice. (C) Representative TEM images of BM MKs in uninfected (i-ii) and infected (iii-iv) mice, showing the presence of multilobulated nucleus (N) and DMS (d) within the cytoplasm. Bar represents 5 μm. Original magnification ×2000. (D) Anti-CD41 mAb-induced ITP in infected and uninfected mice. A single intraperitoneal injection (0.2 µg/g body weight) of anti-CD41 mAb (clone, MWReg30) was administered, followed by collection of blood for platelet counts at the specified time points. Platelet count (E) and rate of decline and recovery of platelet count (F) in uninfected and infected mice after anti-CD41 mAb-induced thrombocytopenia. Data were pooled from 3 independent experiments with uninfected (n = 3), anti-CD41–treated uninfected (n = 9), infected (n = 5), and infected+anti-CD41–treated (n = 10) mice. (G) Representative TEM images of BM MK from infected anti-CD41–treated (i-ii) and uninfected anti-CD41–treated (iii-iv) mice, showing DMS and platelet areas. Bar represents 5 μm. Original magnification ×2000. (H) The perimeter of the demarcation membranes of anti-CD41–treated uninfected (n = 20) and anti-CD41–treated infected (n = 30) mice. The dotted line corresponds with the mean of the perimeter of the DMS in nontreated infected mice. Data are expressed as means ± SEM and analyzed with the unpaired Student t test. *P < .05.
Alteration to BM megakaryocyte form and function during experimental VL. (A) BM MKs were counted manually on hematoxylin and eosin–stained frozen sections in 10 random fields of view for uninfected (n = 4) and infected (n = 4) mice. TEM (FEI Tecnai 12G2) was performed on femur sections of uninfected and day-28–infected mice. Images were analyzed by creating outlines of demarcation membranes with Fiji ImageJ software (supplemental Figure 3). (B) Perimeters were then calculated on 25 uninfected and 46 infected mouse BM MKs. Data represent the mean ± standard error of the mean (SEM) across individual mice. The unpaired Student t test was applied to the means of the perimeter of membranes; ****P < .0001, in uninfected vs infected mice. (C) Representative TEM images of BM MKs in uninfected (i-ii) and infected (iii-iv) mice, showing the presence of multilobulated nucleus (N) and DMS (d) within the cytoplasm. Bar represents 5 μm. Original magnification ×2000. (D) Anti-CD41 mAb-induced ITP in infected and uninfected mice. A single intraperitoneal injection (0.2 µg/g body weight) of anti-CD41 mAb (clone, MWReg30) was administered, followed by collection of blood for platelet counts at the specified time points. Platelet count (E) and rate of decline and recovery of platelet count (F) in uninfected and infected mice after anti-CD41 mAb-induced thrombocytopenia. Data were pooled from 3 independent experiments with uninfected (n = 3), anti-CD41–treated uninfected (n = 9), infected (n = 5), and infected+anti-CD41–treated (n = 10) mice. (G) Representative TEM images of BM MK from infected anti-CD41–treated (i-ii) and uninfected anti-CD41–treated (iii-iv) mice, showing DMS and platelet areas. Bar represents 5 μm. Original magnification ×2000. (H) The perimeter of the demarcation membranes of anti-CD41–treated uninfected (n = 20) and anti-CD41–treated infected (n = 30) mice. The dotted line corresponds with the mean of the perimeter of the DMS in nontreated infected mice. Data are expressed as means ± SEM and analyzed with the unpaired Student t test. *P < .05.
To determine the functional capacity of MKs for platelet production by using an independent approach, we used a well-characterized model of anti-CD41–driven induced ITP.38 Infected and uninfected mice were treated with anti-CD41 mAb and bled for platelet counts at the time points specified in Figure 2D. Administration of anti-CD41 mAb to uninfected and day-28–infected mice led in both cases to a rapid decrease in platelet count followed by a period of recovery to levels above that observed before treatment (Figure 2E). Importantly, the rate of platelet recovery was comparable between groups when normalized to baseline platelet counts (Figure 2F). As restoration in platelet count after experimental ITP is commonly attributed to new platelet production by MKs, these data suggest that the capacity for new platelet production is intact in infected mice, but that extrinsic factors not present in uninfected mice limit the extent to which the platelet count can be restored. A slight reduction in the MPV of anti-CD41–treated infected mice was noted. No difference in the splenic LDUs (146.2 ± 35.07 vs 177.9 ± 54.46) and spleen weights (0.7196 ± 0.304 g vs 0.8298 ± 0.1736 g) were observed between infected anti-CD41–treated and nontreated infected mice. Furthermore, anti-CD41 treatment of infected mice partially restored the integrity of the DMS (17.66 ± 5.68 μm vs 11.84 ± 4.94 μm, anti-CD41–treated infected mice and untreated infected mice, respectively Figure 2H). Although no significant difference in the DMS of uninfected anti-CD41–treated mice was noted, territories of developing platelets were prominent in anti-CD41–treated uninfected and infected mice (Figure 2G).
TPO production is compromised during chronic EVL
Alterations in MK capacity for platelet production may also reflect changes in the availability of TPO, a critical promotor of megakaryopoiesis and platelet production derived largely from hepatocytes.41,42 Given the role of the liver as a target organ in VL, we next assessed the production of TPO during L donovani infection, by measuring circulating unbound TPO levels in uninfected and infected mice. TPO levels in infected mice were significantly reduced at day 28 PI compared with those in uninfected mice (600.527 ± 25.82 pg/mL vs 75.8935 ± 26.75 pg/mL, in uninfected and infected mice respectively; Figure 3A). We also found a 50% reduction in liver Thpo mRNA in infected mice compared with uninfected mice, as determined by quantitative RT-PCR (qRT-PCR; Figure 3B). To better understand the effects of L donovani infection on hepatic TPO production, we stained liver sections of uninfected and day-28–infected mice with anti-TPO antibody (Figure 3C) and analyzed the resulting staining pattern by quantitative morphometry and segmentation analysis. We observed a reduction in the number of TPO+ hepatocytes per unit area, most probably a consequence of granulomatous inflammation; an increase in the number of nonparenchymal inflammatory cells (Figure 3D, left); and an increase in F4/80+ Kupffer cells per unit area (Figure 3D, right). In addition, in liver tissues stained with the anti-TPO antibody and analyzed using an in-house image analysis pipeline, we determined the median fluorescence intensity (MFI) of TPO+ hepatocytes in uninfected vs day-28–infected mice and at different distances (0-30 and 30-60 µm) from the margin of granulomas in the infected mice (supplemental Figure 4). Based on this analysis, TPO expression was marginally but significantly higher in hepatocytes closer (0-30 µm) to granulomas, compared with hepatocytes found farther away (30-60 µm; Figure 3E). Together, these data are suggestive of spatial regulation of TPO production during hepatic infection.
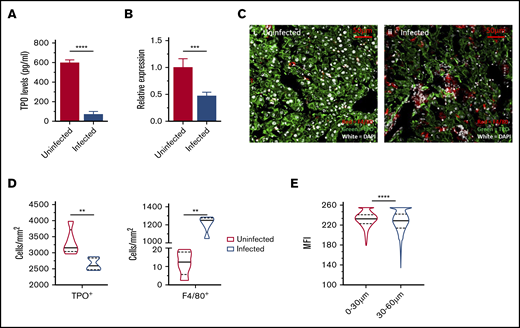
Thrombopoietin regulation during L donovani infection. (A) Circulating levels of TPO at day 28 PI were measured in the serum samples of infected and uninfected C57BL/6 mice. (B) Hepatic mRNA accumulation measured using qRT-PCR on postmortem liver samples of infected and uninfected mice. (C) Representative immunofluorescent liver images of uninfected (i) and infected (ii) mice, stained with anti-TPO antibody (green), F4/80 (red), and 4′,6-diamidino-2-phenylindole (white). Images captured with an LSM 710 confocal microscope at 63× resolution. Bar represents 50 µm. Segmentation analysis was performed with Strataquest image analysis software on immunofluorescent liver images and expressed as cell count per unit area of infected and uninfected livers (D) and MFI of TPO+ cells in infected livers (E). MFIs were calculated for TPO+ hepatocytes at 0 to 30 µm and 30 to 60 µm from the margin of granuloma in infected mice. Data are representative of 5 uninfected and 5 infected mice from a single experiment, analyzed with the unpaired Student t test (A-B) and the nonparametric Mann-Whitney U test (D-E). **P < .01; ***P < .001; ****P < .0001.
Thrombopoietin regulation during L donovani infection. (A) Circulating levels of TPO at day 28 PI were measured in the serum samples of infected and uninfected C57BL/6 mice. (B) Hepatic mRNA accumulation measured using qRT-PCR on postmortem liver samples of infected and uninfected mice. (C) Representative immunofluorescent liver images of uninfected (i) and infected (ii) mice, stained with anti-TPO antibody (green), F4/80 (red), and 4′,6-diamidino-2-phenylindole (white). Images captured with an LSM 710 confocal microscope at 63× resolution. Bar represents 50 µm. Segmentation analysis was performed with Strataquest image analysis software on immunofluorescent liver images and expressed as cell count per unit area of infected and uninfected livers (D) and MFI of TPO+ cells in infected livers (E). MFIs were calculated for TPO+ hepatocytes at 0 to 30 µm and 30 to 60 µm from the margin of granuloma in infected mice. Data are representative of 5 uninfected and 5 infected mice from a single experiment, analyzed with the unpaired Student t test (A-B) and the nonparametric Mann-Whitney U test (D-E). **P < .01; ***P < .001; ****P < .0001.
Splenomegaly-associated platelet clearance in L donovani–infected mice
Although the data indicate significant changes in the host capacity to produce platelets during L donovani infection, changes in reticuloendothelial function, notably associated with splenomegaly, may also have an impact on thrombocytopenia through enhanced platelet clearance. To determine the role of splenomegaly in platelet clearance, sham operated and splenectomized mice were infected with L donovani, and platelet counts were assessed. In control uninfected mice, splenectomy had little impact on platelet count, suggesting that the spleen is not essential for maintaining platelet homeostasis (Figure 4A). In contrast to the pronounced thrombocytopenia observed in sham operated infected mice, splenectomized infected mice were partially protected from thrombocytopenia (P < .001) and had reduced MPV (Figure 4A). We then exposed sham operated and splenectomized infected mice to rTPO to determine the extent to which this cytokine would reconstitute the number of platelets in the absence of a spleen. rTPO administration to sham treated infected mice raised platelet counts by approximately twofold, compared with the range in untreated mice, but this increase remained below the normal range of counts in uninfected mice. These data indicate that rTPO can stimulate platelet production in infected mice, but that there remains a barrier to restoring a homeostatic number of platelets. In contrast, after splenectomy, rTPO elevated the number of platelets in infected mice to supraphysiological levels (Figure 4B). Collectively, these data indicate that, despite the infection-associated changes in MKs noted herein, platelet production is restored by exogenous rTPO and that splenomegaly is a limiting factor governing the number of platelets in the blood.
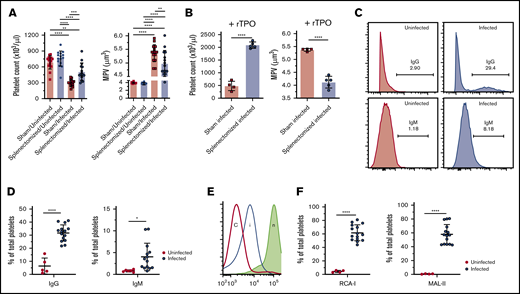
The role of tissue-mediated platelet clearance in L donovani–induced thrombocytopenia. Splenectomized and sham operated mice were infected with L donovani amastigotes for 28 days, similar to WT mice, and bled for the blood counts. (A) Platelet count and MPV of the study groups (n = 15 for each group). Data were pooled from 3 independent experiments and analyzed using ANOVA with the post hoc Tukey’s test comparing mean ± SD of all groups. (B) Platelet count and MPV of sham/infected (n = 4) and splenectomized/infected (n = 5) mice after administration of exogenous rTPO (50 µg/g bodyweight) for 5 consecutive days. Data are representative of a single experiment and analyzed with the unpaired Student t test comparing the mean ± SD between 2 groups. (C) Flow cytometric analysis was performed for the estimation of IgG- and IgM-bound platelets in the circulation of infected mice. Representative histograms show the gating strategy for IgG- and IgM-bound platelets in uninfected and infected mice. (D) Dot plots represent the percentage of total platelets with bound IgG and IgM in uninfected (n = 5) and infected (n = 15) mouse platelets. Data are representative of 2 independent experiments and were analyzed with the unpaired Student t test comparing the mean ± SD between the 2 groups. (E) Flow cytometric analysis was performed on the freshly isolated platelets for the estimation of desialylated platelets in circulation. Histograms representative of gating strategy for lectin+ platelets in infected (i) and uninfected control (c) mice. Neuraminidase-treated platelets (n) were used as the positive control for each lectin. (F) Percentage of RCA-1 and MAL-2 lectin+ platelets from uninfected (n = 4) and infected (n = 15) mice. Data were pooled from 2 independent experiments and analyzed using the unpaired Student t test comparing the mean ± SD between 2 groups. *P < .05; **P < .01; ***P < .001; ****P < .0001.
The role of tissue-mediated platelet clearance in L donovani–induced thrombocytopenia. Splenectomized and sham operated mice were infected with L donovani amastigotes for 28 days, similar to WT mice, and bled for the blood counts. (A) Platelet count and MPV of the study groups (n = 15 for each group). Data were pooled from 3 independent experiments and analyzed using ANOVA with the post hoc Tukey’s test comparing mean ± SD of all groups. (B) Platelet count and MPV of sham/infected (n = 4) and splenectomized/infected (n = 5) mice after administration of exogenous rTPO (50 µg/g bodyweight) for 5 consecutive days. Data are representative of a single experiment and analyzed with the unpaired Student t test comparing the mean ± SD between 2 groups. (C) Flow cytometric analysis was performed for the estimation of IgG- and IgM-bound platelets in the circulation of infected mice. Representative histograms show the gating strategy for IgG- and IgM-bound platelets in uninfected and infected mice. (D) Dot plots represent the percentage of total platelets with bound IgG and IgM in uninfected (n = 5) and infected (n = 15) mouse platelets. Data are representative of 2 independent experiments and were analyzed with the unpaired Student t test comparing the mean ± SD between the 2 groups. (E) Flow cytometric analysis was performed on the freshly isolated platelets for the estimation of desialylated platelets in circulation. Histograms representative of gating strategy for lectin+ platelets in infected (i) and uninfected control (c) mice. Neuraminidase-treated platelets (n) were used as the positive control for each lectin. (F) Percentage of RCA-1 and MAL-2 lectin+ platelets from uninfected (n = 4) and infected (n = 15) mice. Data were pooled from 2 independent experiments and analyzed using the unpaired Student t test comparing the mean ± SD between 2 groups. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Pathways for platelet clearance
Our results indicate that during L donovani infection, there are changes in hepatocyte TPO production and in splenic platelet clearance. To determine the extent to which these events are related to modifications of the platelet membrane and their interaction with host cells, we first determined whether infection is associated with the production of anti-platelet antibodies (Figure 4C). Infected mice showed a significant increase in the frequency of IgG-bound platelets compared with uninfected mice (Figure 4D) and in the amount of IgG bound (MFI 5242.66 ± 811.42 vs 459 ± 312.54; mean ± standard deviation [SD] in infected vs uninfected mice, respectively). Similarly, infected mice had a higher frequency of IgM-bound platelets (Figure 4D) and also bound greater amounts of IgM compared with platelets from uninfected mice (MFI 2068 ± 594.47 vs 420.5 ± 259.50; mean ± SD in infected vs uninfected mice).
Platelet binding to the Ashwell Morrel receptor is associated with desialylation and leads to STAT3 signaling.43 Platelet desialylation results in the exposure of galactose-containing moieties that can be detected using galactose-binding lectins.16 To examine the sialylation status of platelets in infected mice, freshly isolated platelets were stained with 2 galectins: R communis agglutinin-1 (RCA-1) and M amurensis lectin-2 (MAL-2). By flow cytometry, only a low frequency (∼5%) of platelets in uninfected mice were stained with either lectin, as expected from results of previous studies.44,45 In contrast, ∼60% of circulating platelets in infected mice were lectin positive, indicating a marked shift toward desialylation (Figure 4E-F). Thus, L donovani infection is associated with distinct platelet surface changes that facilitate their interaction with both splenic macrophages and hepatocytes.
Posttreatment recovery of platelet count
Clinically and in experimental models of L donovani infection, successful drug treatment can allow for selective and temporal restoration of homeostasis within the immune and hematological compartments.3,46 We therefore sought to examine the extent to which processes that affected platelet count recovered after treatment with AmBisome, a commonly used drug for human VL.47 Platelet counts were monitored in infected mice weekly after AmBisome treatment. Platelet counts (Figure 5A) and MPV (Figure 5B) showed a progressive recovery, reaching normal range within 3 weeks after treatment, with MPV normalizing shortly thereafter. Drug treatment was associated with reduced hepatomegaly, as expected from the resolution of granulomatous inflammation46 (Figure 5C), and that result was accompanied by restoration of serum TPO levels (Figure 5D) and hepatic Thpo mRNA accumulation (Figure 5E). Slow but progressive reduction in splenomegaly was also noted in drug-treated infected mice after treatment (Figure 5F).
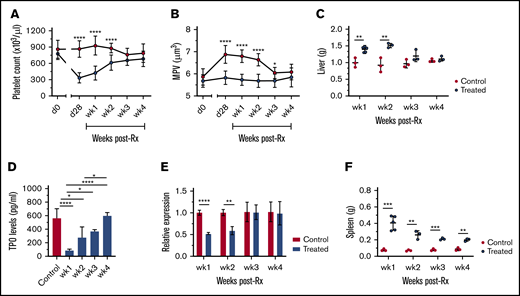
Reversal of thrombocytopenia with the restoration of TPO production and organomegaly in infected mice after treatment.L donovani–infected mice were treated with a single IV dose of AmBisome (8 mg/kg) at day 28 PI and bled weekly via tail vein for platelet counts. Platelet counts (A), MPV (B), and postmortem liver weights (C) of AmBisome-treated and uninfected control mice. Data are representative of a single experiment and were analyzed with the unpaired Student t test comparing means ± SD of treated vs uninfected control at each time point. (D) Circulating TPO levels were measured weekly in serum samples of uninfected control (n = 5) and treated (n = 3 at weeks 1, 2, 3, and 4) mice. Data are representative of a single experiment and analyzed using ANOVA with the post hoc Tukey’s test. (E) Hepatic Thpo mRNA accumulation was measured using quantitative RT-PCR on postmortem liver samples of uninfected control (n = 3) and treated (n = 3) mice at each time point. Data are representative of a single experiment and analyzed using the unpaired Student t test comparing the mean ± SD of control vs treated mice. (F) Postmortem spleen weights (g) of AmBisome-treated and uninfected control mice. Data are representative of a single experiment and analyzed using the unpaired Student t test comparing the mean ± SD of treated vs uninfected control at each time point. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Reversal of thrombocytopenia with the restoration of TPO production and organomegaly in infected mice after treatment.L donovani–infected mice were treated with a single IV dose of AmBisome (8 mg/kg) at day 28 PI and bled weekly via tail vein for platelet counts. Platelet counts (A), MPV (B), and postmortem liver weights (C) of AmBisome-treated and uninfected control mice. Data are representative of a single experiment and were analyzed with the unpaired Student t test comparing means ± SD of treated vs uninfected control at each time point. (D) Circulating TPO levels were measured weekly in serum samples of uninfected control (n = 5) and treated (n = 3 at weeks 1, 2, 3, and 4) mice. Data are representative of a single experiment and analyzed using ANOVA with the post hoc Tukey’s test. (E) Hepatic Thpo mRNA accumulation was measured using quantitative RT-PCR on postmortem liver samples of uninfected control (n = 3) and treated (n = 3) mice at each time point. Data are representative of a single experiment and analyzed using the unpaired Student t test comparing the mean ± SD of control vs treated mice. (F) Postmortem spleen weights (g) of AmBisome-treated and uninfected control mice. Data are representative of a single experiment and analyzed using the unpaired Student t test comparing the mean ± SD of treated vs uninfected control at each time point. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Discussion
Hematological complications including anemia, thrombocytopenia, and pancytopenia have been widely reported in patients with VL and mirrored in many respects in experimental models of the disease,3,25,48 but the underlying mechanisms of thrombocytopenia remain poorly understood. In this study, we showed, in an experimental mouse model of VL, that thrombocytopenia is multifactorial and involves coordinated changes in the platelet surface that lead to both a reduction in production capacity mediated through the TPO/MK axis and increased levels of splenic clearance.
First, we showed that infected C57BL/6 mice develop progressive thrombocytopenia with the presence of giant platelets in circulation. Although the number of BM MKs was similar in control and infected mice, we found that MK maturation was altered in infected mice, leading to clear differences in development of the DMS. Defective MKs with a reduced DMS have been seen in congenital thrombocytopenias, such as MYH9-related disorders,49 but not previously reported in a model of parasitic infection. The demarcation membranes in infected mouse BM MKs were 1.75 times reduced, similar to those observed in congenital MYH9-related disorders.49 Furthermore, we showed that these membranes are recoverable after stimulation of MKs to produce platelets, as evidenced by the increased territories of newly developing platelets after the novel approach of coinduction of ITP with anti-CD41 mAb on the background of chronic L donovani infection. Infected mice showed a pattern of platelet count recovery similar to that in uninfected mice, suggesting that MKs in infected mice retain some functional capability and that other factors contribute to keeping the number of circulating platelets low.
Second, multiple lines of evidence indicated that infection is associated with alteration in TPO production in the liver. Total hepatic mRNA accumulation was decreased, although this may reflect an influx of inflammatory cells leading to dilution of hepatocyte mRNA representation. This finding was supported by a reduction in the number of TPO-expressing hepatocytes detected by immunohistochemistry. Under normal conditions, levels of circulating TPO are inversely proportional to platelet count, although this equilibrium is often dysregulated in hematological and hepatic disease.50-52 In this study, we found a significant decrease in the levels of circulating TPO in infected mice at day 28 PI. Previous studies have shown that the regulation of TPO production is complex and may be independent of both platelet count and circulating levels, as shown by normal levels in ITP and increased levels in aplastic anemia.51 In addition, our results show a reduction in the number of TPO+ hepatocytes, most probably related to hepatic granuloma formation. The expression of TPO, as assessed by MFI, was higher in hepatocytes closer to the margin of granulomas, compared with the hepatocytes more distal to granulomas, suggesting a possible role for cytokines produced by inflammatory cells in the activation of JAK-STAT signaling in hepatocytes.43 Although further studies are needed to confirm a link between form and function of granuloma and hepatocyte production of TPO, our data provide the first indication of spatial regulation of TPO production in the hepatic microenvironment during infection.
Third, we observed that platelets in infected mice were more frequently desialylated than those in uninfected mice. Excessive platelet desialylation has been shown in sepsis and infections that could lead to enhanced platelet clearance by hepatocytes.12,53,54 Although increased platelet desialylation has been proposed to increase the production of TPO by binding to the Ashwell Morrel receptor expressed by hepatocytes,16 this notion has yet to be formally demonstrated in the context of this model of VL. However, disruption of normal hepatic microarchitecture and granulomatous inflammation may alter the signaling pathways for the production of TPO. We have also observed through the application of holotomography that platelets from L donovani–infected mice have additional alterations in their physicochemical properties, such as sphericity, refractive index, and dry mass,55 although it remains to be determined whether these affect the interaction of platelets with hepatocytes.
Finally, for the first time, we have shown in a model of VL the impact of splenectomy on the extent of thrombocytopenia. Splenectomized infected mice had markedly less severe thrombocytopenia than sham operated mice and a better response to exogenous rTPO. Splenomegaly is one of the most common clinical findings in human VL and experimental models. RP macrophages, which act as a major reservoir for the parasite, remain highly phagocytic.37 Macrophage clearance of platelets is often associated with Fc-dependent phagocytosis, and we were also able to demonstrate the presence of antibodies against platelets in circulation. Anti-platelet antibodies have been detected in ITP and infection-induced thrombocytopenia.12 Our data also confirm that platelets are opsonized by anti-platelet IgG and IgM antibodies in murine VL, as previously described in canine VL.26 In this model of infection, mild anemia is also evident from ∼4 weeks PI, but in contrast to thrombocytopenia, anemia is not affected by splenectomy.25 This result is in keeping with the view that, despite both compensatory erythropoiesis and erythrophagocytosis in the spleen,25,56 anemia in EVL is largely driven by CD4+ T-cell–dependent alterations to the BM niche supporting the final stages of erythropoiesis.25
In summary, our findings suggest that the mechanisms responsible for thrombocytopenia are multifactorial, involving both defective BM production and excessive clearance by peripheral tissues. However, these changes appear rapidly and are completely reversible after drug treatment and parasite clearance. These findings provide new insights into the mechanisms and consequences of hematological complications in infections including VL and the management of thrombocytopenia. In addition, we suggest that platelet counts may be a useful surrogate for disease progression or treatment response in both natural infection and experimental models.
Publication-related data and protocols not included in the manuscript will be shared by the authors if requested by e-mail to either corresponding author (ian.hitchcock@york.ac.uk or paul.kaye@york.ac.uk).
Acknowledgments
The authors thank the Biological Services Facility for animal husbandry and the Biosciences Technology Facility Imaging and Cytometry Laboratory for assistance with flow cytometry and imaging; Meg Stark for help with collection of TEM images; and Oliver Herd for help with the TPO enzyme-linked immunosorbent assay.
This work was supported by a Wellcome Trust Senior Investigator Award WT104726 (P.M.K.), British Heart Foundation grant PG/16/26/32099 (I.S.H.), and a scholarship from Khyber Medical University/Higher Education Commission, Pakistan (G.F.R.).
Authorship
Contribution: G.F.R. designed the study, performed the experiments, analyzed the data, and prepared the manuscript; O.P., H.A., and N.B. performed the experiments and analyzed the data; and I.S.H. and P.M.K. oversaw the project, designed the study, prepared the manuscript, and supervised the researchers.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for O.P. is the Institute of Immunity and Transplantation, Royal Free Hospital, University College London, London, United Kingdom.
Correspondence: Ian S. Hitchcock, Department of Biology, York Biomedical Research Institute, University of York, York, YO10 5DD, United Kingdom; e-mail: ian.hitchcock@york.ac.uk; and Paul M. Kaye, York Biomedical Research Institute, Hull York Medical School, University of York, York, YO10 5DD, United Kingdom; e-mail: paul.kaye@york.ac.uk.

