

Key Points
Inhibition of MetAP2 modifies HbS to increase its oxygen affinity and decrease polymerization.
Treatment of sickle cell mice with MetAP2 inhibitors increases red blood cell oxygen affinity and improves ex vivo blood flow.

Abstract
Sickle cell disease (SCD) is associated with hemolysis, vascular inflammation, and organ damage. Affected patients experience chronic painful vaso-occlusive events requiring hospitalization. Hypoxia-induced polymerization of sickle hemoglobin S (HbS) contributes to sickling of red blood cells (RBCs) and disease pathophysiology. Dilution of HbS with nonsickling hemoglobin or hemoglobin with increased oxygen affinity, such as fetal hemoglobin or HbS bound to aromatic aldehydes, is clinically beneficial in decreasing polymerization. We investigated a novel alternate approach to modify HbS and decrease polymerization by inhibiting methionine aminopeptidase 2 (MetAP2), which cleaves the initiator methionine (iMet) from Val1 of α-globin and βS-globin. Kinetic studies with MetAP2 show that βS-globin is a fivefold better substrate than α-globin. Knockdown of MetAP2 in human umbilical cord blood–derived erythroid progenitor 2 cells shows more extensive modification of α-globin than β-globin, consistent with kinetic data. Treatment of human erythroid cells in vitro or Townes SCD mice in vivo with selective MetAP2 inhibitors extensively modifies both globins with N-terminal iMet and acetylated iMet. HbS modification by MetAP2 inhibition increases oxygen affinity, as measured by decreased oxygen tension at which hemoglobin is 50% saturated. Acetyl-iMet modification on βS-globin delays HbS polymerization under hypoxia. MetAP2 inhibitor–treated Townes mice reach 50% total HbS modification, significantly increasing the affinity of RBCs for oxygen, increasing whole blood single-cell RBC oxygen saturation, and decreasing fractional flow velocity losses in blood rheology under decreased oxygen pressures. Crystal structures of modified HbS variants show stabilization of the nonpolymerizing high O2–affinity R2 state, explaining modified HbS antisickling activity. Further study of MetAP2 inhibition as a potential therapeutic target for SCD is warranted.
Introduction
Sickle cell disease (SCD) is the most prevalent genetic hemoglobinopathy worldwide; it affects 330 000 births annually1 and is driven by a point mutation resulting in abnormal βS-globin with Glu6→Val. Hemoglobin S (HbS) polymerizes under low oxygen tension to sickle red blood cells (RBCs), altering blood flow rheology.2 The molecular interactions initiating oligomerization of deoxygenated HbS and mechanism allowing N-terminal modification of α-globin to delay polymerization are supported by structural studies.3,4 A complex SCD pathobiology is dominated by hemolytic anemia and vaso-occlusive events triggered by ischemia and reperfusion with subsequent organ damage and systemic inflammation, endothelial dysfunction, and increased cell-cell binding by adhesion molecules.5-7
Dilution of HbS in RBCs by nonsickling hemoglobin or hemoglobin with increased oxygen affinity, such as fetal hemoglobin (HbF), is clinically beneficial.8-10 For decades, hydroxyurea was the only drug approved by the US Food and Drug Administration for SCD.11 Hydroxyurea induces HbF, which dilutes the fractional concentration of HbS in RBCs and delays hypoxia-induced polymerization. HbF directly destabilizes HbS polymers because it cannot be incorporated into fibers and disrupts intermolecular deoxy-HbS contacts.12 Aldehydes, including 5-hydroxymethylfurfural13 and voxelotor (Oxbryta, GBT-440),14 modify the N terminus of α-globin by reversible covalent imine formation (Schiff base adduct) to increase HbS oxygen affinity (oxygen tension at which hemoglobin is 50% saturated [p50]), allowing HbS to resist polymerization in low oxygen. Voxelotor was approved to treat SCD based on clinical data showing increased hemoglobin levels and improved disease markers, including reduced hemolysis measured by unconjugated bilirubin, reduced reticulocytes, fewer dense RBCs, and a lower percentage of sickled RBCs.15
The current study introduces a novel mechanism for HbS modification by inhibiting the enzyme MetAP2, resulting in reduced HbS polymerization and a concomitant antisickling effect. MetAP2 removes the initiator methionine (iMet) from Val1 in α-globin and β-globin (Figure 1A) as the nascent peptides emerge from the ribosome.16,17 The methionine (Met)-selective Naa50 acetylase subunit of the N-terminal acetyltransferase E complex may then acetylate the retained iMet.18 Covalent fumagillin-derived inhibitors of MetAP2, including TNP-470,19 CKD-732, and ZGN-1061,20 as well as noncovalent reversible inhibitors,21,22 have been described. MetAP2 inhibitors are being developed as chemotherapeutic drugs for antiangiogenic cancer therapy19,22 and for treatment of obesity.20,21 Although the mechanism of action is not fully characterized, retention of iMet by MetAP2 inhibition on eukaryotic elongation factor 1A1 (eEF1A1), a protein with essential translational elongation activity delivering aminoacyl-tRNA to the ribosome, was reported to be a potential cancer therapeutic target and biomarker.22
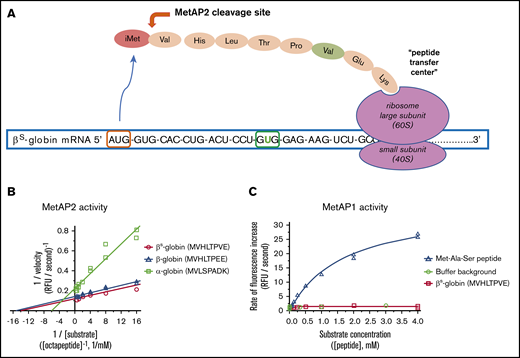
MetAP2 preferentially removes iMet from the N terminus of β-globin, whereas MetAP1 is inactive. (A) MetAP2 is the eukaryotic MetAP that is competent to remove iMet from the N-terminal valine found on both globins as the unfolded globin peptide emerges from the ribosome. Inhibition of MetAP2 prevents iMet removal, allowing N-terminal acetylation by the catalytic Naa50 subunit of the N-terminal acetyltransferase E complex, retaining iMet and acetyl-iMet on a subset of α-globin and β-globin. (B) Lineweaver-Burk plot showing that MetAP2 removes the N-terminal Met from the β-globin and βS-globin octapeptides similarly but fivefold more efficiently compared with the rate of removal of Met from the α-globin N-terminal octapeptide. MetAP2 (9.3 nM) was reconstituted in 30 μM Mn2+, the physiological divalent metal cofactor.54 (C) MetAP1 (57.9 nM) cannot remove Met from the N-terminal βS-globin peptide, but it efficiently cleaves Met from a standard control Met-Ala-Ser tripeptide substrate. MetAP1 was reconstituted with 30 μM Co2+ divalent metal cofactor to maximize activity. MetAP1 and MetAP2 are assayed in a published coupled enzyme assay with L-amino acid oxidase and horseradish peroxidase (Assay 1).55 RFU, relative fluorescent unit.
MetAP2 preferentially removes iMet from the N terminus of β-globin, whereas MetAP1 is inactive. (A) MetAP2 is the eukaryotic MetAP that is competent to remove iMet from the N-terminal valine found on both globins as the unfolded globin peptide emerges from the ribosome. Inhibition of MetAP2 prevents iMet removal, allowing N-terminal acetylation by the catalytic Naa50 subunit of the N-terminal acetyltransferase E complex, retaining iMet and acetyl-iMet on a subset of α-globin and β-globin. (B) Lineweaver-Burk plot showing that MetAP2 removes the N-terminal Met from the β-globin and βS-globin octapeptides similarly but fivefold more efficiently compared with the rate of removal of Met from the α-globin N-terminal octapeptide. MetAP2 (9.3 nM) was reconstituted in 30 μM Mn2+, the physiological divalent metal cofactor.54 (C) MetAP1 (57.9 nM) cannot remove Met from the N-terminal βS-globin peptide, but it efficiently cleaves Met from a standard control Met-Ala-Ser tripeptide substrate. MetAP1 was reconstituted with 30 μM Co2+ divalent metal cofactor to maximize activity. MetAP1 and MetAP2 are assayed in a published coupled enzyme assay with L-amino acid oxidase and horseradish peroxidase (Assay 1).55 RFU, relative fluorescent unit.
We tested the hypothesis that inhibition of MetAP2 results in irreversible modification of HbS by N-terminal retention of iMet and subsequent acetylation to acetylated iMet (acetyl-iMet). These N-terminal modifications of HbS stabilize the high O2–affinity R2 state of modified HbS, increase its oxygen affinity to delay polymerization, and prevent RBC sickling under low oxygen tension.
Methods
Analysis of N-terminal modified globin by LC-MS
RBCs or differentiated erythroid cells (∼106 cells) were washed twice with phosphate-buffered saline, lysed in 50 µL of high-performance liquid chromatography–grade water by freeze/thaw, vortexed, and centrifuged (10 000g, 5 minutes, 4°C) before exchanging any residual buffer in the supernatant with water using a Zeba Spin Desalting Column, 7K MWCO, 0.5 mL (Thermo Fisher Scientific, Waltham, MA). Samples were transferred into new containers and stored at −80°C prior to analysis. Intact globin masses determined by liquid chromatography-mass spectrometry (LC-MS) through use of a Waters ACQUITY BEH300 C4 reverse phase column (2.1 × 100 mm, 300 Å pore, 1.7 μm; part no. 186004496) on an Agilent 1290 Infinity II Ultra High Performance Liquid Chromatographer coupled to an Agilent 6530 Accurate Mass Electrospray Ionization Quadrupole Time-of-Flight mass spectrometer. Spectra were deconvoluted to calculate globin modification percentages.
Crystal structure determination of modified HbS samples
The following purified samples of modified HbS were used for crystallization and structure determination: (1) acetyl-iMet–β-globin (96.1%) with iMet–α-globin (86.6%) and unmodified α-globin (11.2%), referred to as CRYST 1, and (2) unmodified β-globin (93%) with a mixture of α-globins (19% unmodified α-globin, 58% iMet–α-globin, and 23% acetyl-iMet–α-globin), referred to as CRYST 2 (supplemental Methods).
Study approvals
Whole blood was collected from adult (>18 years) SCD donors at steady-state, who had not received a blood transfusion within 3 months, in a heparin Vacutainer tube under a Western Institutional Review Board–approved protocol (WIRB #1138926), following informed consent prior to the blood draw. In vivo experiments with mice conformed to the guidelines of the Institutional Animal Care and Use Committees of Bioverativ/Sanofi. Generation of MetAP2 His231→Asn mice was performed at Applied StemCell (Milpitas, CA), following protocol guidelines from the Institutional Animal Care and Use Committees.
Results
MetAP2 removes iMet fivefold more efficiently from the β-globin N terminus
All 3 α-globin, β-globin, and βS-globin N-terminal octapeptides are good substrates for MetAP2 compared with a standard tripeptide substrate Met-Ala-Ser in Mn2+ buffer. Kinetic parameters for Met removal from β-globin and βS-globin octapeptides were determined, including the maximun rate (Vmax) and the Michaelis constant (Km), and both are similar. However, Met is cleaved from β-globin peptides with fivefold greater catalytic efficiency compared with the α-globin peptide, as shown by the improved Vmax/Km (Figure 1B; Table 1). As anticipated from published substrate specificity,23 MetAP1 efficiently processes the Met-Ala-Ser tripeptide but cannot process the βS-globin octapeptide (Figure 1C). MetAP2 cleavage of the βS-globin peptide was compared with the human eEF1A1 N-terminal peptide.22 A 4.7-fold lower Km for βS-globin octapeptide indicates that βS-globin is better recognized by MetAP2 than by eEF1A1 in low substrate concentrations. However, a higher Vmax for cleavage of eEF1A1 octapeptide results in a twofold improvement in Vmax/Km catalytic efficiency for cleavage of eEF1A1 (Table 1).
MetAP2 enzyme kinetic studies for Met cleavage from N-terminal peptides
| Peptide | Source | Km, mM | Vmax, RFU/s | Vmax/Km, RFU · L/s · mmol |
|---|---|---|---|---|
| Assay 1* | ||||
| MAS | Tripeptide substrate | 0.53 | 21 | 39 |
| MVHLTPVE | βS-globin N terminus | 0.086 | 8.7 | 102 |
| MVHLTPEE | β-globin N terminus | 0.074 | 7.2 | 98 |
| MVLSPADK | α-globin N terminus | 0.23 | 5.0 | 21 |
| Assay 2† | ||||
| MVHLTPVE | βS-globin N terminus | 0.019 | 0.011 | 0.55 |
| MGKGKTHI | Human eEF1A1 | 0.089 | 0.097 | 1.1 |
| Peptide | Source | Km, mM | Vmax, RFU/s | Vmax/Km, RFU · L/s · mmol |
|---|---|---|---|---|
| Assay 1* | ||||
| MAS | Tripeptide substrate | 0.53 | 21 | 39 |
| MVHLTPVE | βS-globin N terminus | 0.086 | 8.7 | 102 |
| MVHLTPEE | β-globin N terminus | 0.074 | 7.2 | 98 |
| MVLSPADK | α-globin N terminus | 0.23 | 5.0 | 21 |
| Assay 2† | ||||
| MVHLTPVE | βS-globin N terminus | 0.019 | 0.011 | 0.55 |
| MGKGKTHI | Human eEF1A1 | 0.089 | 0.097 | 1.1 |
Human MetAP2 was assayed in buffer containing 30 µM Mn2+, the physiological cofactor divalent cation,53 using the indicated coupling enzymes to detect released Met.
A, alanine; D, aspartate; E, glutamate; H, histidine; K, lysine; L, leucine; M, methionine; P, proline; S, serine; T, threonine, V, valine.
Assay 1 = 9.3 nM MetAP2, amino acid oxidase, O2, horseradish peroxidase, Amplex Red. Tris(2-carboxyethyl)phosphine is not included because of coupling enzyme–dependent background interference.
Assay 2 = 2.5 nM MetAP2, leucine dehydrogenase, NAD+.
High levels of MetAP2 knockdown or inhibition are needed to modify HbS in vitro
Decreased protein expression of MetAP2 was confirmed in a transfected short hairpin RNA (shRNA) human umbilical cord blood–derived erythroid progenitor 2 (HUDEP-2) cell pool (supplemental Methods) by immunohistochemical staining of permeabilized cells or western blot analysis of cell lysates and modification of α-globin by iMet and acetyl-iMet confirmed by LC-MS. MetAP2 expression in individual subclones of the pool varied from 79% to ∼1% (limit of detection) by western blot analysis. No baseline globin modification is observed in wild-type HUDEP-2 cells by LC-MS (Figure 2A). Total modification of α-globin by iMet and acetyl-iMet in individual subclones correlates with the decrease in MetAP2 by western blot analysis (supplemental Table 3). Clone H3, with the greatest decrease in MetAP2 (∼99%), allows retention of iMet and acetyl-iMet on both globins (Figure 2B); however α-globin is more highly modified (>50%) than β-globin (<10%), consistent with enzyme kinetic studies of N-terminal Met removal.
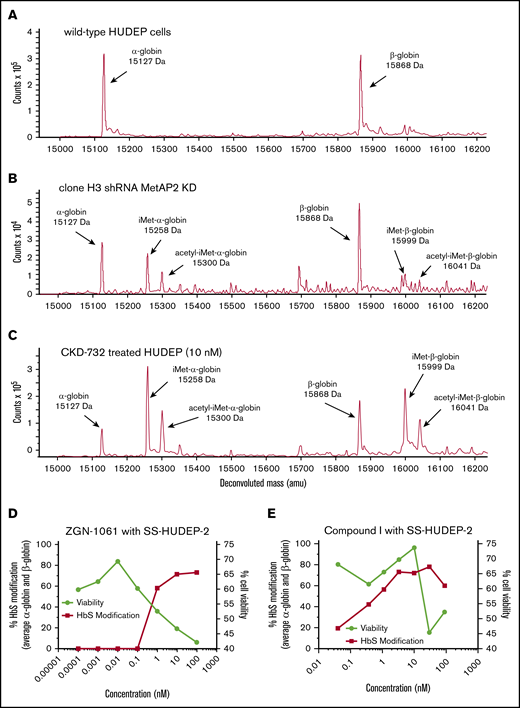
Globin modification in HUDEP-2 and SS-HUDEP-2 cells by MetAP2 knockdown or inhibition. The N termini of α-globin and β-globin are modified with iMet and acetyl-iMet in wild-type HUDEP-2 cells treated with lentivirus expressing MetAP2-specific shRNA or in HUDEP-2 and SS-HUDEP-224 cells treated with MetAP2 inhibitors. (A) Analysis by LC-MS confirms that wild-type HUDEP-2 cells do not have baseline globin modification. (B) Globin modification in subclone H3 from a pool of lentivirus-treated HUDEP-2 cells. MetAP2 expression in clone H3 is decreased ∼99% based on western blot analysis (supplemental Table 3). Because of the smaller sample size, nonglobin background peaks are more evident in this sample. (C) Treatment of HUDEP-2 cells during differentiation with the selective covalent MetAP2 inhibitor CKD-732 (10 nM) leads to high levels of HbA modification (∼80%) on both globins (85% α-globin and 74% β-globin modification). Similar results were obtained in 2 independent studies. (D) SS-HUDEP-2 cells were differentiated for 7 days in the indicated concentrations of the covalent, irreversible MetAP2 inhibitor ZGN-1061,20 to increase modification of HbS by iMet retention at concentrations that decrease cell viability (88% α-globin and 59.5% βS-globin maximum modification). (E) In contrast, a reversible inhibitor of MetAP2, Compound I,21 increases HbS modification at concentrations that do not affect cell viability (93.5% α-globin and 63% βS-globin maximum modification). Similar results were obtained in 2 independent studies.
Globin modification in HUDEP-2 and SS-HUDEP-2 cells by MetAP2 knockdown or inhibition. The N termini of α-globin and β-globin are modified with iMet and acetyl-iMet in wild-type HUDEP-2 cells treated with lentivirus expressing MetAP2-specific shRNA or in HUDEP-2 and SS-HUDEP-224 cells treated with MetAP2 inhibitors. (A) Analysis by LC-MS confirms that wild-type HUDEP-2 cells do not have baseline globin modification. (B) Globin modification in subclone H3 from a pool of lentivirus-treated HUDEP-2 cells. MetAP2 expression in clone H3 is decreased ∼99% based on western blot analysis (supplemental Table 3). Because of the smaller sample size, nonglobin background peaks are more evident in this sample. (C) Treatment of HUDEP-2 cells during differentiation with the selective covalent MetAP2 inhibitor CKD-732 (10 nM) leads to high levels of HbA modification (∼80%) on both globins (85% α-globin and 74% β-globin modification). Similar results were obtained in 2 independent studies. (D) SS-HUDEP-2 cells were differentiated for 7 days in the indicated concentrations of the covalent, irreversible MetAP2 inhibitor ZGN-1061,20 to increase modification of HbS by iMet retention at concentrations that decrease cell viability (88% α-globin and 59.5% βS-globin maximum modification). (E) In contrast, a reversible inhibitor of MetAP2, Compound I,21 increases HbS modification at concentrations that do not affect cell viability (93.5% α-globin and 63% βS-globin maximum modification). Similar results were obtained in 2 independent studies.
HUDEP-2 cells treated during differentiation with a selective covalent MetAP2 inhibitor CKD-73220 resulted in extensive globin modification (∼75% total HbS; Figure 2C). Covalent fumagillin-like inhibitors and reversible MetAP2 inhibitors are active in erythroid cells in culture. The dose response for the covalent inhibitor ZGN-106120 and a reversible competitive MetAP2 inhibitor Compound I21 was compared in sickle cell HUDEP-2 (SS-HUDEP-2) cells containing the Glu6→Val βS-globin mutation.24 The covalent inhibitor is more potent, but covalent and reversible MetAP2 inhibitors achieve extensive HbS modification (∼75%). The dose response for ZGN-1061 is steep, and globin modification occurs only at inhibitor concentrations that decrease cell viability (Figure 2D). In contrast, Compound I with a normal sigmoidal dose response curve achieves maximal globin modification at concentrations (3.3 and 10 µM) that do not affect cell viability, although viability decreases at higher concentrations (30 and 100 µM; Figure 2E). HbS modification was also observed in differentiating peripheral blood mononuclear cell–derived erythroid cells from SCD donor blood treated for 7 days with 10 μM Compound I, reaching 59% α-globin and 29% βS-globin modification without affecting cell viability.
MetAP2 knockout and inhibition in humanized SCD Townes mice
We replaced wild-type mouse MetAP2 with an active site His231→Asn mutant (supplemental Methods) to ablate MetAP2 activity while retaining expression of the MetAP2/p67 protein to regulate eIF2α phosphorylation.25 Transgenic SCD mice expressing the MetAP2 His231→Asn mutant are embryonically lethal by embryonic day 8.5, a phenotype resembling the complete MetAP2 knockout.26 RBCs from heterozygous mice expressing ∼50% MetAP2 protein (supplemental Figure 3) do not show any N-terminal HbS modification by iMet or acetyl-iMet, indicating that a much higher loss of MetAP2 activity is necessary to achieve extensive HbS modification.
Treatment of SCD mice with covalent MetAP2 inhibitors CKD-732, ZGN-1061, or TNP-470 results in extensive HbS modification. Vehicle-treated Townes mice have no detectible N-terminal globin modification (Figure 3A). A pilot study, using TNP-470, 30 mg/kg injected intraperitoneally every third day for 3 weeks (7 injections; n = 5), results in 47% total HbS modification (51% α-globin modification and 40% βS-globin modification). Subsequent studies used lower daily doses of less toxic fumagillin analogs. Treatment with intraperitoneal CKD-732, 3 mg/kg daily for 3 days, results in 50% total HbS modification: 59% α-globin modification (41% iMet–α-globin and 18% acetyl-iMet–α-globin) and 41.5% βS-globin (27% iMet–βS-globin and 14.5% acetyl-iMet–βS-globin) (Figure 3B). Increasing the dose to 10 or 30 mg/kg or extending daily treatment with 3 mg/kg for 14 days results in the same 50% modification of HbS; however, 2-week daily dosing with 10 or 30 mg/kg results in toxicity, including lethargy, weight loss, and deaths at the highest dose (data not shown). Decreasing the daily dose of CKD-732 to 1 mg/kg decreases modification (37% α-globin and 23.5% βS-globin). Treatment with intraperitoneal ZGN-1061, 1 mg/kg daily for 3 days, results in similar HbS modification as CKD-732 (41% α-globin and 27.5% βS-globin). Extending the daily dosing to 45 days does not increase maximum HbS modification (43% α-globin and 27.5% βS-globin). High levels of HbS modification are maintained in these chronic dosing studies with no observed toxicity; however, chronic treatment does not increase RBC levels (supplemental Figure 6). Daily treatment with ZGN-1061, 0.3 mg/kg, an efficacious dose for weight loss in diet-induced obese mice,20 results in lower total HbS modification (34% α-globin and 22.5% βS-globin). Townes mice treated with ZGN-1061, 0.3 and 1 mg/kg, gain weight similarly to vehicle controls over 45 days. Although treatment of SS-HUDEP-2 cells with Compound I resulted in high HbS modification (Figure 2E), extended treatment in SCD mice, 10 mg/kg intraperitoneally daily, results in modest HbS modification (17% α-globin and 3% βS-globin), despite peak plasma drug level values of >10 μM. However, these peak plasma levels are not sustained, even with twice-daily dosing, as a result of the rapid clearance of Compound I in mice (data not shown). Covalent inhibitors were used in all subsequent studies in vivo.
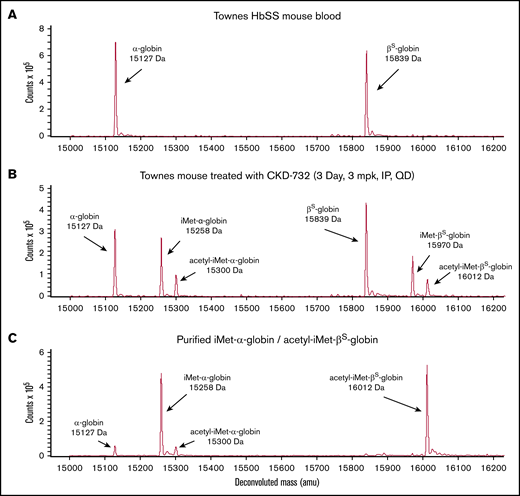
Modification of human HbS by iMet and acetyl-iMet in MetAP2 inhibitor treated Townes SCD mice. The N termini of α-globin (15 127 Da) and βS-globin (15 839 Da) are modified with iMet (15 258 Da and 15 970 Da, respectively) or acetyl-iMet (15 300 Da and 16 012 Da, respectively) in Townes SCD mice treated with MetAP2 inhibitor. (A) LC-MS analysis of RBC lysates from Townes mice treated with vehicle does not show baseline globin modification. (B) Townes mice treated intraperitoneally with CKD-732, 3 mg/kg daily for 3 days, show extensive modification of α-globin and β-globin (50% total HbS modification). Similar results were obtained in 5 independent studies. (C) Repeated cation-exchange chromatography yields highly pure samples of modified HbS for x-ray crystallographic structure studies with a single modification on each globin. Sample shown is acetyl-iMet–βS-globin (96.1%) and iMet–α-globin (86.6%).
Modification of human HbS by iMet and acetyl-iMet in MetAP2 inhibitor treated Townes SCD mice. The N termini of α-globin (15 127 Da) and βS-globin (15 839 Da) are modified with iMet (15 258 Da and 15 970 Da, respectively) or acetyl-iMet (15 300 Da and 16 012 Da, respectively) in Townes SCD mice treated with MetAP2 inhibitor. (A) LC-MS analysis of RBC lysates from Townes mice treated with vehicle does not show baseline globin modification. (B) Townes mice treated intraperitoneally with CKD-732, 3 mg/kg daily for 3 days, show extensive modification of α-globin and β-globin (50% total HbS modification). Similar results were obtained in 5 independent studies. (C) Repeated cation-exchange chromatography yields highly pure samples of modified HbS for x-ray crystallographic structure studies with a single modification on each globin. Sample shown is acetyl-iMet–βS-globin (96.1%) and iMet–α-globin (86.6%).
Characterization of iMet- and acetyl-iMet–modified HbS purified from Townes mice
Modified HbS was purified from RBCs collected from Townes mice treated with covalent MetAP2 inhibitors (supplemental Methods). Nine possible combinations of modified α-globin and βS-globin in the HbS tetramer were separated into 3 major pools (supplemental Figure 4) dominated by the modifications on the βS-globin (unmodified βS-globin, iMet–βS-globin, and acetyl-iMet–βS-globin) with unresolved combinations of modified and unmodified α-globin. Modified HbS purified for crystallization included a sample of 96.1% acetyl-iMet–βS-globin and 86.8% iMet–α-globin (Figure 3C). Acetyl-iMet–βS-globin combinations represent only 10% to 15% of total HbS in treated mice; therefore, 3 pools of modified HbS were compared with unmodified HbS in biochemical assays without further fractionation because of sample limitations. The composition of the modified and unmodified globins in the 3 pools is shown in Table 2.
Percentage decrease in oxygen saturation p50 relative to unmodified HbS
| Globin analysis in sample | 2,3-BPG | |||
|---|---|---|---|---|
| βS-globin | α-globin | 5.0 mM | 9.7 mM | 14.2 mM |
| acetyl-iMet- | acetyl-iMet-/iMet-/unmodified (1:2:1) | 38% | nd* | nd |
| iMet- | acetyl-iMet-/iMet-/unmodified (1:3:1) | 14%, 12% | 12% | 12% |
| Unmodified | acetyl-iMet-/iMet- (1:1) | 10%, 5% | 16.5% | 25% |
| Globin analysis in sample | 2,3-BPG | |||
|---|---|---|---|---|
| βS-globin | α-globin | 5.0 mM | 9.7 mM | 14.2 mM |
| acetyl-iMet- | acetyl-iMet-/iMet-/unmodified (1:2:1) | 38% | nd* | nd |
| iMet- | acetyl-iMet-/iMet-/unmodified (1:3:1) | 14%, 12% | 12% | 12% |
| Unmodified | acetyl-iMet-/iMet- (1:1) | 10%, 5% | 16.5% | 25% |
nd, not done.
Due to the limited amount of protein, only 1 concentration of 2,3-BPG was tested.
All 3 modified HbS pools bind oxygen with higher affinity than does unmodified HbS. Oxygen affinity of purified HbS samples was determined in the presence of 5 mM 2,3-bisphosphoglycerate (2,3-BPG), and p50 values were calculated from the oxygen equilibrium curves (OECs) (Figure 4A). Under these conditions, the p50 of pure hemoglobin is lower than the p50 of intact RBCs. The p50 of unmodified HbS is 21.1 torr; the p50 of unmodified βS-globin with a mixture of acetyl-iMet–α-globin and iMet–α-globin is 19.0 torr (10% increased O2 affinity); the p50 of iMet–βS-globin with a mixture of acetyl-iMet–α-globin, iMet–α-globin, and unmodified α-globin is 18.2 torr (14% increase); and the p50 of acetyl-iMet–βS-globin with a mixture of acetyl-iMet–α-globin, iMet–α-globin and unmodified α-globin is 13.1 torr (38% increase). The increased oxygen affinity compared with unmodified HbS in several 2,3-BPG concentrations ranges from 10% to 38% for the modified HbS pools (Table 2). In addition to increasing oxygen affinity, acetyl-iMet–βS-globin modification delays HbS polymerization under hypoxia. Retention of acetyl-iMet on the βS-globin chain of HbS delays polymerization in 1.895 M potassium phosphate buffer27 under conditions of complete hypoxia created by the oxygen scavenger metabisulfite. Polymerization times are delayed by 10% to 20% for the acetyl-iMet–βS-globin HbS pool relative to unmodified HbS in contrast to the other modified pools (Figure 4B-C). Dilution of purified acetyl-iMet–βS-globin into pure HbS significantly delays polymerization of the bulk hemoglobin mixture starting at 30% acetyl-iMet–βS-globin and 70% HbS (Figure 4D).
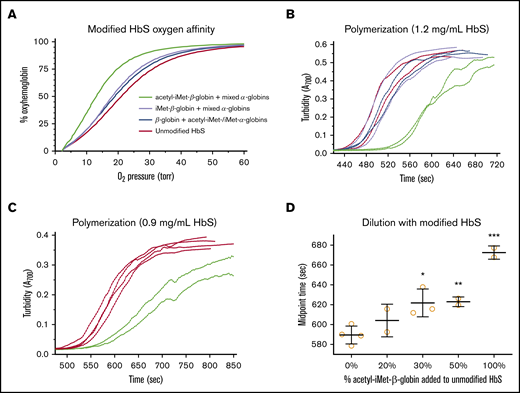
Modification of HbS with iMet and acetyl-iMet increases oxygen affinity and delays polymerization. Purified samples of modified HbS (Table 2; supplemental Figure 4) were tested for oxygen binding and polymerization under complete hypoxia. (A) Oxygen affinity determined from OECs in the presence of 5 mM 2,3-BPG at 37°C. 2,3-BPG stabilizes deoxyhemoglobin and is added to mimic levels in RBCs. OEC of unmodified HbS (red line, 21.1 torr) shows that p50 for pure HbS is lower than p50 measured in intact RBCs in whole blood (∼28 torr; Figure 5D). Modified HbS samples have higher affinities for oxygen, as shown by OEC for modified HbS containing unmodified βS-globin with a mixture of acetyl-iMet–α-globin and iMet–α-globin (blue line, 19.0 torr), OEC for iMet–βS-globin with a mixture of acetyl-iMet–α-globin, iMet–α-globin, and unmodified α-globin (purple line, 18.2 torr), and OEC for acetyl-iMet–βS-globin with a mixture of acetyl-iMet–α-globin, iMet–α-globin, and unmodified α-globin (green line, 13.1 torr). (B) Acetyl-iMet–βS-globin modification of HbS delays polymerization under hypoxia, as shown by turbidity increases at 700 nm of samples assayed at 1.2 mg/mL in 1% sodium metabisulfite and 1.895 M potassium phosphate, pH 7.5, in sealed cuvettes at 37°C. The delay time for acetyl-iMet–βS-globin with a mixture of acetyl-iMet–α-globin, iMet–α-globin, and unmodified α-globin (green lines) is increased by 80 seconds (17%), and the midpoint time is increased by 100 seconds (20%) relative to HbS (red lines). Polymerization of unmodified βS-globin with a mixture of acetyl-iMet–α-globin and iMet–α-globin (blue lines) or iMet–βS-globin with a mixture of acetyl-iMet–α-globin, iMet–α-globin, and unmodified α-globin (purple lines) are similar to unmodified HbS. Similar results were obtained in 2 independent studies. (C) For 0.9 mg/mL HbS samples, the delay times of acetyl-iMet–βS-globin with the mixture of α-globins (green lines) is increased by 54 seconds (10%) and the midpoint time increased by 83 seconds (14%) relative to HbS (red lines). (D) Dilution of purified acetyl-iMet–βS-globin into pure HbS to a final concentration of 0.9 mg/mL significantly delays polymerization of the bulk hemoglobin mixture starting at 30% acetyl-iMet–βS-globin and 70% HbS. The delay time to initiate turbidity increase and the time to reach the midpoint of maximal turbidity were determined using a modified high phosphate buffer assay of HbS polymerization.27 ***P < .005, **P < .01, *P < .05, 2-tailed Student t test.
Modification of HbS with iMet and acetyl-iMet increases oxygen affinity and delays polymerization. Purified samples of modified HbS (Table 2; supplemental Figure 4) were tested for oxygen binding and polymerization under complete hypoxia. (A) Oxygen affinity determined from OECs in the presence of 5 mM 2,3-BPG at 37°C. 2,3-BPG stabilizes deoxyhemoglobin and is added to mimic levels in RBCs. OEC of unmodified HbS (red line, 21.1 torr) shows that p50 for pure HbS is lower than p50 measured in intact RBCs in whole blood (∼28 torr; Figure 5D). Modified HbS samples have higher affinities for oxygen, as shown by OEC for modified HbS containing unmodified βS-globin with a mixture of acetyl-iMet–α-globin and iMet–α-globin (blue line, 19.0 torr), OEC for iMet–βS-globin with a mixture of acetyl-iMet–α-globin, iMet–α-globin, and unmodified α-globin (purple line, 18.2 torr), and OEC for acetyl-iMet–βS-globin with a mixture of acetyl-iMet–α-globin, iMet–α-globin, and unmodified α-globin (green line, 13.1 torr). (B) Acetyl-iMet–βS-globin modification of HbS delays polymerization under hypoxia, as shown by turbidity increases at 700 nm of samples assayed at 1.2 mg/mL in 1% sodium metabisulfite and 1.895 M potassium phosphate, pH 7.5, in sealed cuvettes at 37°C. The delay time for acetyl-iMet–βS-globin with a mixture of acetyl-iMet–α-globin, iMet–α-globin, and unmodified α-globin (green lines) is increased by 80 seconds (17%), and the midpoint time is increased by 100 seconds (20%) relative to HbS (red lines). Polymerization of unmodified βS-globin with a mixture of acetyl-iMet–α-globin and iMet–α-globin (blue lines) or iMet–βS-globin with a mixture of acetyl-iMet–α-globin, iMet–α-globin, and unmodified α-globin (purple lines) are similar to unmodified HbS. Similar results were obtained in 2 independent studies. (C) For 0.9 mg/mL HbS samples, the delay times of acetyl-iMet–βS-globin with the mixture of α-globins (green lines) is increased by 54 seconds (10%) and the midpoint time increased by 83 seconds (14%) relative to HbS (red lines). (D) Dilution of purified acetyl-iMet–βS-globin into pure HbS to a final concentration of 0.9 mg/mL significantly delays polymerization of the bulk hemoglobin mixture starting at 30% acetyl-iMet–βS-globin and 70% HbS. The delay time to initiate turbidity increase and the time to reach the midpoint of maximal turbidity were determined using a modified high phosphate buffer assay of HbS polymerization.27 ***P < .005, **P < .01, *P < .05, 2-tailed Student t test.
Inhibition of MetAP2 modifies HbS to decrease RBC sickling, increase oxygen saturation, and improve blood flow rheology
Biochemical improvements observed with purified N-terminal modified HbS translated into hematological benefits in blood from SCD mice treated with MetAP2 inhibitors in assays of RBC sickling, oxygen binding, and blood flow rheology. Time course studies with TNP-470, CKD-732, and ZGN-1061 all reach maximal HbS modification (∼50%) after 3 days; therefore, mice were dosed daily for 3 days intraperitoneally with vehicle or CKD-732 (10 mg/kg) to achieve 50% total HbS modification (59% α-globin and 41.5% βS-globin). A published sickle cell imaging flow cytometry assay (SIFCA)28 was performed on whole blood incubated under normoxia (21% oxygen) or hypoxia (4% oxygen) for 2.5 hours prior to fixing with 2.5% EM-grade glutaraldehyde to quantitate abnormal sickled cells. Treatment with MetAP2 inhibitor results in a 58% decrease in hypoxia-driven RBC sickling by SIFCA (Figure 5A) and a 34% decrease by imaging fixed cells on the PerkinElmer Operetta CLS high-content analysis system (Figure 5B). We attempted to increase HbS modification > 50%, by dosing CKD-732 and ZGN-1061 twice daily at 1 mg/kg (Figure 5C), and found total modification is similar for both (51% total HbS modification, 55% α-globin, and 46.5% βS-globin), reaching the same modification as a single daily 10 mg/kg dose. Oxygen affinity (p50) determined in whole blood collected in EDTA from Townes mice treated with ZGN-1061 increased by 3.43 ± 0.24 torr (mean ± standard deviation) or 12.2% vs vehicle-control blood (Figure 5D).

Modification of HbS by MetAP2 inhibition decreases hypoxia-triggered sickling and increases oxygen binding in whole blood RBCs. Townes SCD mice were dosed intraperitoneally for 3 days with the indicated covalent MetAP2 inhibitor or vehicle control, and whole blood was collected to measure HbS modification, hypoxia-triggered RBC sickling, and oxygen affinity (p50). (A) Whole blood from SCD mice dosed with CKD-732 (10 mg/kg, daily) to achieve 50% total HbS modification was evaluated for sickling under hypoxia by SIFCA,29 performed under normoxia (21% oxygen) or hypoxia (4% oxygen) for 2 to 3 hours prior to fixing and quantitating abnormal sickled cells on the Amnis ImageStream Imaging Flow Cytometer. (B) Sickling was evaluated on a PerkinElmer Operetta CLS High-Content Analysis System. Both methods confirm a decrease in hypoxia-driven sickling in whole blood samples from MetAP2 inhibitor–treated mice. Similar results were obtained in ≥3 independent studies for both methods. (C) SCD mice dosed with the indicated covalent MetAP2 inhibitors (1 mg/kg, twice daily) achieved similar high levels of HbS modification, as assessed by loss of the parent HbS peak on a Bio-Rad D-10 Hemoglobin Testing System, consistent with LC-MS analysis of lysed RBCs showing 55% total α-globin modification and 46.5% β-globin modification. (D) Oxygen affinity (p50) determined from RBC OECs in whole blood collected in EDTA. The p50 measured for MetAP2 inhibitor–treated blood samples is significantly lower, and oxygen affinity increased by 3.43 ± 0.24 torr (mean ± standard deviation) or 12.2% compared with vehicle controls. Similar results were obtained in 3 independent studies. ****P < .001, ***P < .005, 2-tailed Student t test. ns, not significant.
Modification of HbS by MetAP2 inhibition decreases hypoxia-triggered sickling and increases oxygen binding in whole blood RBCs. Townes SCD mice were dosed intraperitoneally for 3 days with the indicated covalent MetAP2 inhibitor or vehicle control, and whole blood was collected to measure HbS modification, hypoxia-triggered RBC sickling, and oxygen affinity (p50). (A) Whole blood from SCD mice dosed with CKD-732 (10 mg/kg, daily) to achieve 50% total HbS modification was evaluated for sickling under hypoxia by SIFCA,29 performed under normoxia (21% oxygen) or hypoxia (4% oxygen) for 2 to 3 hours prior to fixing and quantitating abnormal sickled cells on the Amnis ImageStream Imaging Flow Cytometer. (B) Sickling was evaluated on a PerkinElmer Operetta CLS High-Content Analysis System. Both methods confirm a decrease in hypoxia-driven sickling in whole blood samples from MetAP2 inhibitor–treated mice. Similar results were obtained in ≥3 independent studies for both methods. (C) SCD mice dosed with the indicated covalent MetAP2 inhibitors (1 mg/kg, twice daily) achieved similar high levels of HbS modification, as assessed by loss of the parent HbS peak on a Bio-Rad D-10 Hemoglobin Testing System, consistent with LC-MS analysis of lysed RBCs showing 55% total α-globin modification and 46.5% β-globin modification. (D) Oxygen affinity (p50) determined from RBC OECs in whole blood collected in EDTA. The p50 measured for MetAP2 inhibitor–treated blood samples is significantly lower, and oxygen affinity increased by 3.43 ± 0.24 torr (mean ± standard deviation) or 12.2% compared with vehicle controls. Similar results were obtained in 3 independent studies. ****P < .001, ***P < .005, 2-tailed Student t test. ns, not significant.
Pooled blood from CKD-732– or ZGN-1061–treated SCD mice (1 mg/kg, twice daily) was compared with untreated controls in an assay that measures single-cell oxygen saturation as a function of the percentage of oxygen.29 Modification of HbS allows higher levels of HbS saturation at lower oxygen levels compared with untreated blood (Figure 6A; supplemental Table 5). Comparison of the median percentage of oxygen saturation values of control blood with CKD-732–treated mouse blood shows improved oxygen saturation at all oxygen partial pressures (Figure 6B). Individual mouse samples were tested to determine whether treatment improves blood flow rheology in low oxygen. Samples from mice treated with CKD-732 (3 days, 1 mg/kg, intraperitoneally, twice daily) and control mice were tested in an assay of oxygen-dependent blood flow rheology.30,31 In MetAP2 inhibitor-treated samples containing modified HbS, the oxygen threshold needed to affect lower blood flow significantly is decreased (Figure 6C), and the total blood flow velocity drop when transitioning from 21% to 0% oxygen is significantly lower (Figure 6D).
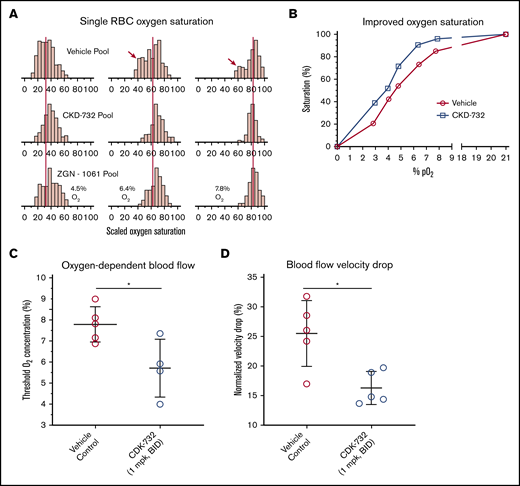
Improvements in RBC oxygen saturation and flow rheology under low oxygen in blood from SCD mice treated with MetAP2 inhibitor. Pooled whole blood from Townes SCD mice treated for 3 days intraperitoneally with CKD-732 or ZGN-1061 (1 mg/kg, twice daily) or vehicle control (n = 5 in each pool) were compared. (A) Single-cell oxygen saturation as a function of oxygen partial pressure was determined,29 and frequency histograms of 2 treated pools compared with the vehicle pool for 4.5%, 6.4%, and 7.8% O2. The red vertical lines are placed on the median percentage of oxygen saturation values of the vehicle pool and show a similar shift by both MetAP2 inhibitors to higher levels of saturation. Red arrows indicate the bimodal distribution of oxygen saturation, including a population of cells with low oxygen saturation that is absent in the treated pools (complete histograms for all pO2 values, including hypoxia and normoxia controls, are shown as supplemental Figure 5). (B) Median percentage of oxygen saturation values for the vehicle pool compared with the CKD-732–treated pool shows improved oxygen saturation at all oxygen partial pressures. Similar results were obtained in 2 independent studies. (C) Individual blood samples from mice treated with CKD-732 and controls were tested in an assay of oxygen-dependent blood flow rheology.30,31 Oxygen threshold to lower blood flow is significantly decreased in MetAP2 inhibitor–treated blood samples containing modified HbS. *P = .026, 2-tailed Student t test. (D) Total observed blood flow velocity drop, when transitioning from 21% to 0% oxygen, is significantly lower. *P = .011, 2-tailed Student t test. Similar results were obtained in 2 independent studies.
Improvements in RBC oxygen saturation and flow rheology under low oxygen in blood from SCD mice treated with MetAP2 inhibitor. Pooled whole blood from Townes SCD mice treated for 3 days intraperitoneally with CKD-732 or ZGN-1061 (1 mg/kg, twice daily) or vehicle control (n = 5 in each pool) were compared. (A) Single-cell oxygen saturation as a function of oxygen partial pressure was determined,29 and frequency histograms of 2 treated pools compared with the vehicle pool for 4.5%, 6.4%, and 7.8% O2. The red vertical lines are placed on the median percentage of oxygen saturation values of the vehicle pool and show a similar shift by both MetAP2 inhibitors to higher levels of saturation. Red arrows indicate the bimodal distribution of oxygen saturation, including a population of cells with low oxygen saturation that is absent in the treated pools (complete histograms for all pO2 values, including hypoxia and normoxia controls, are shown as supplemental Figure 5). (B) Median percentage of oxygen saturation values for the vehicle pool compared with the CKD-732–treated pool shows improved oxygen saturation at all oxygen partial pressures. Similar results were obtained in 2 independent studies. (C) Individual blood samples from mice treated with CKD-732 and controls were tested in an assay of oxygen-dependent blood flow rheology.30,31 Oxygen threshold to lower blood flow is significantly decreased in MetAP2 inhibitor–treated blood samples containing modified HbS. *P = .026, 2-tailed Student t test. (D) Total observed blood flow velocity drop, when transitioning from 21% to 0% oxygen, is significantly lower. *P = .011, 2-tailed Student t test. Similar results were obtained in 2 independent studies.
Crystallographic studies of modified HbS proteins
Two N-terminal modified HbS variants were purified and crystallized in the CO-ligated forms for structural studies. CRYST 1 is acetyl-iMet–β-globin (96.1%), iMet–α-globin (86.6%), and unmodified α-globin (11.2%), whereas CRYST 2 is unmodified β-globin (93%), unmodified α-globin (19%), iMet–α-globin (58%), and acetyl-iMet–α-globin (23%). Both crystals have 5 molecules of αβ-dimers in the asymmetric unit and in the R2 state previously described for structures of aldehyde-modified hemoglobin.4,13,32 We observed significant disorder in some of the globin chains, explaining the high Rfactor/Rfree of the CRYST 1 and CRYST 2 structures (23.9/32.3% and 26.3/36.4%, respectively). The disorder in CRYST 2 prevented structure refinement to completion. Nonetheless, at Rfree of 36%, we observed apparent α-globin chain modifications as described above, clearly indicating that modification of α-globin alone is able to stabilize the relaxed structure, akin to aromatic-aldehyde–modified R2 hemoglobin. Detailed crystallographic and refinement parameters for CRYST 1 are reported in Table 3.
Detailed crystallographic and refinement parameters
| Parameter | Value |
|---|---|
| Data collection statistics | |
| Space group | I 1 2 1 |
| Unit-cell a, b, c (Å, degree) | 134.84, 53.85, 195.64, 90, 91.73, 90 |
| Resolution, Å | 29.07-2.5 (2.59-2.50) |
| Unique reflections, n | 49 161 (4856) |
| Redundancy | 4.4 (4.4) |
| Completeness, % | 99.93 (99.79) |
| Average I/σ(I) | 13.62 (1.80) |
| Rmerge, %* | 8.3 (53.0) |
| Refinement statistics | |
| Resolution, Å | 29.07-2.50 |
| No. of reflections | 49 061 |
| Rwork, % | 23.87 (29.46) |
| Rfree, %† | 32.28 (42.61) |
| R.m.s.d. bonds, Å | 0.008 |
| R.m.s.d. angles, degree | 1.360 |
| Dihedral angles, % | |
| Most favored | 91.61 |
| Allowed | 6.83 |
| Average B (Å2)/atoms | |
| All atoms | 54.80 |
| Macromolecule | 55.20 |
| Ligands | 51.80 |
| Water | 47.30 |
| Parameter | Value |
|---|---|
| Data collection statistics | |
| Space group | I 1 2 1 |
| Unit-cell a, b, c (Å, degree) | 134.84, 53.85, 195.64, 90, 91.73, 90 |
| Resolution, Å | 29.07-2.5 (2.59-2.50) |
| Unique reflections, n | 49 161 (4856) |
| Redundancy | 4.4 (4.4) |
| Completeness, % | 99.93 (99.79) |
| Average I/σ(I) | 13.62 (1.80) |
| Rmerge, %* | 8.3 (53.0) |
| Refinement statistics | |
| Resolution, Å | 29.07-2.50 |
| No. of reflections | 49 061 |
| Rwork, % | 23.87 (29.46) |
| Rfree, %† | 32.28 (42.61) |
| R.m.s.d. bonds, Å | 0.008 |
| R.m.s.d. angles, degree | 1.360 |
| Dihedral angles, % | |
| Most favored | 91.61 |
| Allowed | 6.83 |
| Average B (Å2)/atoms | |
| All atoms | 54.80 |
| Macromolecule | 55.20 |
| Ligands | 51.80 |
| Water | 47.30 |
Rfree and Rwork are measures of the quality between the crystallographic model and experimental X-ray diffraction data. R.m.s.d., root mean square deviation of atomic positions.
Rmerge = ΣhklΣi|Ii(hkl) − < I(hkl)>|/ΣhklΣiIi(hkl).
Rfree was calculated from 5% randomly selected reflection for cross-validation. All other measured reflections were used during refinement.
The crystal structure of CRYST 1 (acetyl-iMet–βS-globin with iMet–α-globin) shows that the 2 retained iMet nearly fill the α-cleft, making close hydrophobic interactions with residues from the same α-globin and the opposite α-globin chains (α1Thr134, α1Ser131, α1Lys127, α2Thr134, and α2Thr138) (Figure 7A-B). These interactions are expected to stabilize the relaxed state conformation and increase the oxygen affinity of the variant HbS. Interestingly, comparison of the modified variant HbS structure with the aromatic aldehyde TD-7 modified hemoglobin structure (PDB code 6D14) shows the 2 covalently bound iMet residues at the α-cleft superimpose closely on the covalently bound TD-7 compounds (Figure 7C).32 In a manner reminiscent of aromatic aldehyde-hemoglobin adducts, the 2 iMet residues on the α-globin chains are too bulky to occupy the α-cleft of the classical R state.4 As expected for liganded hemoglobin in the R2 state conformation, the 2 acetyl-iMet residues in the β-cleft of CRYST 1 are relatively far apart (Figure 7D). Like the iMet at the α-globin, the acetyl-iMet also makes close hydrophobic/hydrophilic interactions with residues from the same and opposite β-globin chains (β1His146, β1Tyr145, β2Asn139, β2Gly136, β2Ala135, β2Leu78, β2Asn80, and β2Asp79) (Figure 7E) that also contribute to the relaxed state stabilization and increase the variant hemoglobin affinity for oxygen.

X-ray crystal structure of acetyl-iMet–βS-globin with iMet–α-globin modified HbS (CRYST 1) showing altered α-cleft and β-cleft. The CO-ligated structure of the modified HbS is in the R2 state previously described for structures of aldehyde modified HbS.4,32 (A) Structure of acetyl-iMet–βS-globin with iMet-α–globin modified HbS shows that the 2 iMet residues (cyan spheres) at the α-globin chains nearly fill the α-cleft. (B) Close interactions of iMet (contoured with final 2Fo-Fc electron density at 0.8σ) with residues from α1- and α2-globin chains. (C) The α-cleft region in the structure of acetyl-iMet–βS-globin with iMet–α-globin (cyan) is compared with the same region of a structure of HbS modified by the covalently bound antisickling aromatic aldehyde TD-7 (magenta). (D) The β-cleft of acetyl-iMet–βS-globin with iMet–α-globin is shown with the acetyl-iMet residues highlighted (magenta). (E) Close interactions of acetyl-iMet (contoured with final 2Fo-Fc electron density at 0.6σ) with residues from the β1- and β2-globin chains.
X-ray crystal structure of acetyl-iMet–βS-globin with iMet–α-globin modified HbS (CRYST 1) showing altered α-cleft and β-cleft. The CO-ligated structure of the modified HbS is in the R2 state previously described for structures of aldehyde modified HbS.4,32 (A) Structure of acetyl-iMet–βS-globin with iMet-α–globin modified HbS shows that the 2 iMet residues (cyan spheres) at the α-globin chains nearly fill the α-cleft. (B) Close interactions of iMet (contoured with final 2Fo-Fc electron density at 0.8σ) with residues from α1- and α2-globin chains. (C) The α-cleft region in the structure of acetyl-iMet–βS-globin with iMet–α-globin (cyan) is compared with the same region of a structure of HbS modified by the covalently bound antisickling aromatic aldehyde TD-7 (magenta). (D) The β-cleft of acetyl-iMet–βS-globin with iMet–α-globin is shown with the acetyl-iMet residues highlighted (magenta). (E) Close interactions of acetyl-iMet (contoured with final 2Fo-Fc electron density at 0.6σ) with residues from the β1- and β2-globin chains.
Discussion
Inhibition of MetAP2 results in retention of iMet on α-globin and β-globin, allowing additional partial acetylation of the retained iMet to create a mixture of HbS tetramers modified on the N termini of both globins (unmodified, iMet globin, and acetyl-iMet globin). MetAP2 inhibition allows modification of 50% total HbS in SCD mice in vivo and 75% total HbS in human SS-HUDEP-2 cells24 in vitro. Modified HbS variants resist polymerization and RBC sickling under conditions of low O2 by delaying HbS polymerization and increasing O2 affinity. In blood from SCD mice treated with MetAP2 inhibitors, RBCs are less sickled, with a higher affinity for oxygen binding, and oxygen-dependent blood flow rheology in low O2 is improved. Notably, the increased RBC oxygen affinity reported here (12.5% decrease in p50) is similar in magnitude to decreases achieved by alternate antisickling approaches. Treatment with the pyruvate kinase-R activator FT-4202 decreases p50 in SCD mice by 7%,33 and similar decreases are reported in the clinic.34 Our data suggest that MetAP2 may warrant further study as a potential therapeutic target for SCD.
Clinical testing of the covalent MetAP2 inhibitor CKD-732 to treat obesity in Prader-Willi syndrome decreased hyperphagia and resulted in weight loss. However, serious venous thrombotic adverse events were observed in CKD-732–treated participants, with 2 fatal events of pulmonary embolism and 2 events of deep vein thrombosis compared with placebo.35 It is not reported whether on-target mechanisms link MetAP2 inhibition to thrombosis or whether the reactive epoxide in this covalent fumagillin-like inhibitor triggers off-target effects. In the current study, we found that a noncovalent MetAP2 inhibitor is not cytotoxic at efficacious concentrations for modification of HbS in SS-HUDEP-2 erythroid cells compared with cytotoxicity observed with a covalent MetAP2 inhibitor in vitro. Covalent MetAP2 inhibitors CDK-732 and ZGN-1061 are efficacious in weight loss studies in obese mice.20 We found that even higher doses of ZGN-1061 than were used in a diet-induced obesity mouse model do not cause weight loss in chronic studies in SCD mice, perhaps reflecting altered tissue expression of MetAP2 reported in diet-induced obese mice.36 However, chronic dosing of covalent MetAP2 inhibitors in SCD did not increase RBCs, despite improvements to RBC oxygen affinity and sickling. Perhaps these beneficial effects on RBCs are offset by cytotoxic effects on erythroid cells, as observed in vitro. Further studies are needed to determine the potential for safe on-target inhibition of MetAP2 in erythroid cells in vivo.
At baseline, SCD mice have no detectible level of N-terminal globin modification. Daily treatment with covalent MetAP2 inhibitors for only 3 days results in a maximal 50% total HbS modification. Extending treatment to twice daily or longer duration does not increase the level of modification. This rapid response to MetAP2 inhibition is likely due to the rapid turnover of RBCs in SCD mice. RBC half-life is 2.5 days in homozygous HbSS Townes mice in contrast to 16 days in HbAA control mice,37 and 56.8% of circulating RBCs are reticulocytes.38 Rapid attainment of maximal HbS modification is likely a combination of accelerated erythropoiesis and the ability of CKD-732 to inhibit MetAP2 in circulating reticulocytes that synthesize HbS after release from bone marrow.
Mammalian cells express 3 MetAPs, posttranslational modification enzymes operating at the peptide transfer center of the ribosome.16,17 MetAP1, MetAP1D, and MetAP2 all remove iMet from small side chain amino acids (Gly, Pro, Ala, Ser, Cys), but MetAP2 also removes iMet from Thr and Val with slightly larger side chains. Enzyme kinetic studies with MetAP1 and MetAP2 are consistent with our observations in erythroid cells in vitro and in RBCs in vivo. Only MetAP2 removes N-terminal Met from β-globin, βS-globin, and α-globin N-terminal octapeptides, in agreement with known substrate specificity.23 We confirmed that MetAP1 cannot remove N-terminal Met from the βS-globin octapeptide. N-terminal octapeptides used to measure MetAP2 activity39 represent good surrogates for physiological substrates that are unfolded as they emerge from the 60S subunit.16,17 The catalytic efficiencies for removal of Met from β-globin and βS-globin octapeptides are very similar, reflecting the fact that substitution of Glu6 for Val6 is far removed from the N-terminal iMet. However, the efficiency of Met removal from α-globin is fivefold lower than β-globin, consistent with the observation that inhibition of MetAP2 favors iMet retention on α-globin in erythroid cells in vitro and in SCD mice in vivo. Improved efficiency of iMet cleavage from β-globin results in greater retention on α-globin, because residual MetAP2 removes iMet from β-globin over α-globin.
The role of MetAP2 in erythroid cell differentiation is complex. Removal of iMet and N-terminal acetylation are common posttranslational modifications17 potentially resulting in profound changes to protein function, as exemplified by studies with KRAS.40 However, removal of iMet from the N terminus of α-globin or β-globin is nonessential for oxygen delivery by hemoglobin. Many hemoglobin variants are characterized by mutations near their N termini that result in retention of iMet, with or without subsequent acetylation to acetyl-iMet. These mutations, including α-globin41,42 and β-globin43-46 N-terminal mutations, do not affect oxygen delivery or cause anemia. Although iMet removal from hemoglobin does not affect oxygen delivery, iMet removal may be important to conserve the essential amino acid Met only obtained in the diet of mammals. Failure to remove iMet from the 4 N termini of 2 mM hemoglobin would result in an additional 8 mM bound Met in blood.
The MetAP2 protein has evolved a second important function in erythroid cells as a regulator of protein synthesis. During erythropoiesis, heme-regulated eukaryotic elongation factor 2α (eIF2α) kinase or heme-regulated inhibitor (HRI) coordinates translation of α-globin and β-globin messenger RNA to match the availability of cellular heme. Active HRI, lacking heme bound in the regulatory site, phosphorylates the eIF2α domain (eIF2α) subunit of eIF2 complex to inhibit eIF2 initiation of ribosomal messenger RNA translation. HRI is inhibited when heme levels increase, allowing globin synthesis. HRI expression increases during erythroid differentiation,47 reaching high levels when hemoglobin synthesis is maximal.48 Heme addition to inhibit HRI maximizes in vitro translation in rabbit reticulocyte lysates, and p67 is a protein factor that decreases the inhibition of initiation by HRI.49,50 MetAP2 and p67 are identical,51 and MetAP2 copurifies with eIF2, inhibiting phosphorylation of eIF2α.25 We show that expression of MetAP2 also increases during erythroid differentiation, similar to increases reported for HRI.48 Increases in MetAP2 during erythroid differentiation may regulate the activity of increasing levels of HRI. Alternately, MetAP2 may increase to facilitate cleavage of high iMet-globin levels during terminal differentiation.
When MetAP2 is knocked down in HUDEP-2 cells by shRNA, clones that retain expression of ≤55% MetAP2 protein have a maximum of 21% α-globin modification, the globin most easily modified by MetAP2 inhibition. A clone with 99% loss of MetAP2 protein only resulted in 55% α-globin modification, suggesting a high loss of MetAP2 activity is needed to retain iMet on hemoglobin. Inhibition of MetAP2 in SS-HUDEP-2 cells expressing βS-globin by covalent or reversible inhibitors achieves ∼75% total HbS modification, whereas MetAP2 inhibition in mice reaches 50% total HbS modification with covalent inhibitors. These differences between inhibition in vitro and in vivo may be due to the rapid turnover of bone marrow erythroid cells in SCD mice, as reflected by the 2.5-day RBC half-life,37 combined with high in vivo clearance of MetAP2 inhibitors.20 Alternately, high-level HbS modification by covalent inhibitors may be limited by cell toxicity.
Events leading to the eponymous shape change in sickle RBCs that triggers SCD pathology are complex and include HbS oligomerization, polymerization, secondary nucleation of fibers, fiber branching, and growth resulting in rigid sickle RBCs.52 All are initiated by dimerization of 2 deoxy-HbS tetramers through the interaction of mutant βVal6 in the βS-globin of 1 tetramer with a hydrophobic pocket (Ala70, Phe85, Leu88) in a βS-globin in a second tetramer, as found in the deoxy-HbS structure.3 We were unable to crystallize modified deoxy-HbS directly to compare the effect of N-terminal iMet retention and acetylation on the nearby hydrophobic interaction triggered by Val6; however, oxy-HbS structures of CO-liganded modified HbS variants provide insight into the mechanism by which MetAP2 inhibition increases the affinity of modified HbS for oxygen and decreases polymerization.
The structures of both modified HbS variants (βS-globin with iMet–α-globin and acetyl-iMet–βS-globin with iMet–α-globin) are in the R2 state conformation previously described for structures of aldehyde modified hemoglobin.4,13,32 The R2 state is 1 of several high O2–affinity relaxed-state ensembles, including classical R, RR2, R3, and RR3, and is the only relaxed conformation capable of binding antisickling aromatic aldehydes at the α-cleft.4,53 The 2 retained iMet residues nearly fill the α-cleft in the CO-ligated structure of acetyl-iMet–βS-globin/iMet–α-globin modified HbS, similarly to the structure observed with a bound antisickling aromatic aldehyde (Figure 7C). Like aromatic aldehydes, interactions between the iMet residues and α-cleft residues stabilize the relaxed state. The crystal structure of βS-globin/iMet–α-globin modified HbS is also in the R2 state, indicating that α-globin cleft modification alone suffices to stabilize the R2 state. In the R2 state, the 2 βVal1 residues are further apart compared with the T state, in which the β-globin Val1 residues are much closer to each other. Not surprisingly, modification of the βS-globin Val1 residues with acetyl-iMet in the acetyl-iMet–βS-globin/iMet–α-globin structure shows that the acetyl-iMet are far apart; nonetheless, acetyl-iMet interacts with both βS-globin chains to stabilize the relaxed state conformation and increase the modified hemoglobin oxygen affinity. In the narrowed β-cleft of modified deoxy-HbS, these acetyl-iMet residues may sterically affect binding of 2,3-BPG, providing a direct mechanism for increasing oxygen affinity by reversing the stabilizing effect of 2,3-BGP binding to deoxy-HbS.
In summary, retention of iMet and subsequent acetylation on α-globin and βS-globin improve erythrocytes in SCD blood by increasing their oxygen affinity, decreasing their tendency to sickle under hypoxia, increasing single-cell oxygen saturation, and decreasing the blood flow velocity loss in low oxygen. As a potential therapeutic target to treat SCD, erythrocyte MetAP2 must be inhibited safely and sufficiently to ensure a high level of HbS modification. Erythroid-specific inhibitors of MetAP2 with limited distribution into other tissues may allow safe modification of HbS with benefit in SCD.
Data sharing requests should be sent to David R. Light (davidrlight@gmail.com).
Acknowledgments
The authors thank their colleagues at Bioverativ and Sanofi, including Neelan Sivaneri and Reid Williams for optimizing MetAP enzyme assays, Vu Hong for discussions during the development of Assay 2, Alejandra Macias-Garcia for dosing animal models, Carolina Morell-Perez for p50 determinations, Arjan van der Flier for discussions about testing the His231→Asn MetAP2 mice, Sungtaek Lim for discussions of MetAP2 inhibitors, Hong Sheng for compound purification, Andre Bourque for stability analysis of SPEW formulations, Heather Davis for compound formulation support, Qi Lu for discussions of eEF1A1, and Sriram Krishnamoorthy, and Courtney Mercadante for advice on the manuscript. They also thank Yukio Nakamura (RIKEN BioResource Research Center, Koyadai, Tsukuba, Japan) for providing the HUDEP-2 erythroid cell line and James A. Wells (University of California San Francisco) for his original suggestion to investigate posttranslational modification of HbS.
Structural biology resources were provided by National Institutes of Health Shared Instrumentation Grant S10OD021756 (M.K.S.) and Virginia General Assembly Higher Education Equipment Trust Fund to Virginia Commonwealth University (M.K.S.).
Authorship
Contribution: M.D. and S.S. designed and analyzed knockdown and erythroid cell treatment studies; M.D., S.S., K.R.G., and A.H. designed and analyzed SCD mouse dosing studies; M.D. and D.G. designed and characterized the SS-HUDEP-2 cells; D.R.L. and K.D. designed and analyzed enzyme kinetic studies; B.F.V., D.G., and K.D. designed and analyzed RBC sickling and oxygen affinity studies in blood; W.L. analyzed and characterized the mutant MetAP2 mouse model; A.I. and D.R.L. designed and analyzed studies to purify modified HbS samples; B.P.G., G.D.C., E.S., and J.M.H. designed and analyzed single RBC oxygen saturation studies; S.H. and D.K.W. designed and analyzed the sickle mouse blood rheology studies; F.N.M. and M.K.S. designed and analyzed modified HbS structural studies; and D.R.L., M.K.S., A.H., and M.D. wrote and revised the manuscript.
Conflict-of-interest disclosure: M.D., S.S., K.R.G., A.H., B.F.V., D.G., K.D., A.I., W.L., and D.R.L. are employees of Bioverativ, a subsidiary of Sanofi, and have equity ownership in the company. M.K.S., J.M.H., and D.K.W. received research funding from Bioverativ, a subsidiary of Sanofi. The remaining authors declare no competing financial interests.
Correspondence: David R. Light, Sanofi, Rare Blood Disorders, 225 2nd Ave, Waltham, MA 02451; e-mail: davidrlight@gmail.com.

