

TO THE EDITOR:
To better understand platelet-leukocyte interactions and identify new molecular interactions that play a role in thrombo-inflammatory disease, we performed a systematic protein interaction screen of 122 platelet and 128 leukocyte cell surface receptors and secreted proteins using an AVidity-based EXtracellular Interaction Screen (AVEXIS). This screen was developed to identify low-affinity interactions between receptors and their ligands, such as those that exist between membrane proteins.1,2 The basis of the screen is the generation of monomeric and pentameric protein complexes (bait and prey proteins) and panning using β-lactamase activity of prey proteins (Figure 1A). The screen primarily focuses on single transmembrane proteins with a few other structural classes of extracellular proteins. Multispanning transmembrane receptors are largely excluded because they cannot be expressed as functional proteins using conventional techniques.
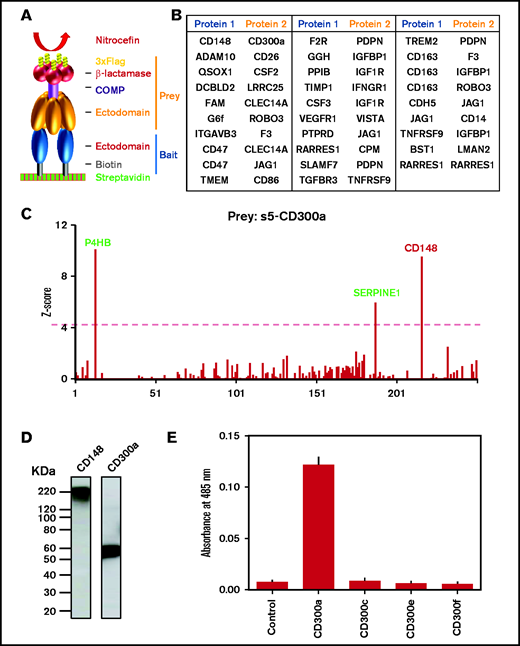
AVEXIS screen identified a potential novel interaction between CD300a and CD148 receptors. (A) Normalized monobiotinylated bait proteins were arrayed, incubated with pentameric preys, washed, and detected with β-lactamase substrate (nitrocefin) using AVEXIS. (B) List of novel interactions identified in the current study. Complete information is shown in supplemental Table 3. (C) Prey s5-CD300a was screened against 250 monomeric bait proteins from our human platelet-leukocyte extracellular protein library; it detected CD148 as a positive hit. Two other promiscuous baits proteins also appeared in this screen (green text). The red dashed line represents the Z-score cutoff of 4. (D) Western blots showing expression of CD148 and CD300a bait proteins with expected molecular weights. (E) Prey CD300 family receptors and a negative control protein were screened against CD148 bait; only CD300a showed a positive interaction (n ≥ 3). COMP, cartilage oligomeric matrix protein.
AVEXIS screen identified a potential novel interaction between CD300a and CD148 receptors. (A) Normalized monobiotinylated bait proteins were arrayed, incubated with pentameric preys, washed, and detected with β-lactamase substrate (nitrocefin) using AVEXIS. (B) List of novel interactions identified in the current study. Complete information is shown in supplemental Table 3. (C) Prey s5-CD300a was screened against 250 monomeric bait proteins from our human platelet-leukocyte extracellular protein library; it detected CD148 as a positive hit. Two other promiscuous baits proteins also appeared in this screen (green text). The red dashed line represents the Z-score cutoff of 4. (D) Western blots showing expression of CD148 and CD300a bait proteins with expected molecular weights. (E) Prey CD300 family receptors and a negative control protein were screened against CD148 bait; only CD300a showed a positive interaction (n ≥ 3). COMP, cartilage oligomeric matrix protein.
The monocyte receptors and secreted proteins were identified by analyzing 16 published proteomic data sets describing the proteome of monocytes and monocyte-derived macrophages using a suite of bioinformatics tools. To increase the chances of identifying functionally relevant interactions, care was taken to preserve the extracellular binding function by expressing the proteins in 5 structural categories readily expressed using current methodologies and using different cloning constructs: type I/GPI-anchored proteins (n = 119), type II (n = 13), secreted (n = 15), heterodimeric complexes (n = 8), and multispan transmembrane proteins (n = 5).3 A total of 176 plasmids encoding 160 proteins and their complexes were synthesized (some proteins are encoded by multiple subunits) and expressed in mammalian cells (supplemental Table 1) to ensure that structurally important posttranslational modifications, such as disulfide bonds and glycans, were added. After purification, 122 platelet proteins that we described previously3 and 128 monocyte proteins (supplemental Table 1) were successfully expressed as monomeric biotinylated “baits” for screening. ProteinSimple western blots of the protein library showed that the vast majority (99/109; 91%) of proteins exhibited mass heterogeneity centered on their expected size, suggesting the presence of different glycoforms (supplemental Figure 1). We were able to express 80 of the proteins (12 platelets, 68 monocytes) as pentameric prey proteins at levels sufficient for interaction screening (supplemental Table 1). The recombinant protein library represents a valuable resource for the investigation of human platelet and monocyte biology.
To systematically identify novel receptor-ligand interactions involved in human platelet-monocyte interactions, we screened our monocyte receptor prey library with the platelet and monocyte baits using the AVEXIS assay (supplemental Figure 2). In total, 19 369 potential direct binary interactions were tested. Positive interacting baits for each prey were initially identified as described in supplemental Methods. In total, 36 interactions between platelet and monocyte/macrophage proteins were identified (Figure 1B; supplemental Tables 2 and 3). Seven of the receptor-ligand pairs were previously known, including between CLEC-2 and podoplanin4 and CSF2 and its receptors CSF2RA and CSF2RB,5 whereas the remaining 29 interactions (Figure 1B) are novel. The novel interactions included ones between CLEC14A and CD47 or FAM (Figure 1B; supplemental Figure 3). CLEC14A is expressed on endothelial cells and has been found to interact with multimerin-2, which regulates angiogenesis and tumor progression.6 It is not known whether CD47 or FAM share the same binding region with multimerin-2 or whether they also regulate vessel formation. CD47 is a critical regulator of myeloid cell activation and serves a broader role as a myeloid-specific immune checkpoint; it is of interest to investigate whether this is regulated by CLEC14A.
A novel interaction between the leukocyte receptor CD300a and CD148 was identified, along with 2 other promiscuous baits (defined as interacting with >10% of the preys tested): P4HB and SERPINE13 (Figure 1C). CD148 is the only known transmembrane tyrosine phosphatase in platelets, and it regulates immunoreceptor tyrosine-based activation motif (ITAM) receptor signaling.7 The endogenous ligand for CD148 is unknown. Loss-of-function genetic variants of CD148 from human patients caused autosomal-recessive thrombocytopenia.8
CD300a is an immunoreceptor tyrosine-based inhibitory motif receptor that is expressed in leukocyte populations9,10 and involved in inflammatory cytokine and chemokine production.11 CD300a−/− mice show increased neutrophil recruitment, enhanced bacterial clearance, and prolonged survival after cecal ligation and puncture.11 Hypoxia, a common feature of a proinflammatory response, can induce CD300a expression in human monocytes and macrophages.12 CD300a is also upregulated on monocytes during transendothelial migration.
We performed a validation screen to verify the interaction and to investigate whether CD148 binds to other members of the CD300 family. The result shows that CD300a is the only member that interacts with CD148, including the closely related family member, CD300c (Figure 1E). Because the most divergent region between CD300a and CD300c is toward the C-terminal membrane proximal region of the ectodomain, we speculate that this region is involved in CD300a binding to CD148.
We performed experiments to investigate the functional significance of the interaction between CD300a and CD148. Platelets incubated with the pentameric CD300a (s5-CD300a), not a control protein s5-CD200, exhibited increased tyrosine phosphorylation of several proteins with a prominent band at 72 kDa and several smaller bands, including at 34 kDa and >150 kDa (Figure 2A). This included increased phosphorylation of Src family kinases on their excitatory tyrosine residue in the catalytic domain (Figure 2A).

CD300a promotes signaling and inhibits platelet aggregations via CD148. (A) s5-CD300a (40 nM) increases global phosphorylated tyrosine (p-Tyr) in unactivated platelets in the presence of secondary mediator inhibitors (n = 3). (B) s5-CD300a (40 nM, 200 nM for monomer) inhibits rhodocytin-induced (50 nM) platelet aggregation in humans and mice, which is abolished by anti-human/mouse CD148 antibody (10 µg/mL) (n ≥ 3). (C) s5-CD300a, not a control protein s5-CD200, inhibits Src and Lat phosphorylation by rhodocytin (n = 3). (D) s5-CD300a (40 nM, 200 nM for monomer) inhibits rhodocytin-induced (50 nM) platelet aggregation in human platelet-rich plasma. (E) s5-CD148 induces CD300a tyrosine phosphorylation in CD300a-transfected HEK293T cells. IP, immunoprecipitation; ′′, minutes.
CD300a promotes signaling and inhibits platelet aggregations via CD148. (A) s5-CD300a (40 nM) increases global phosphorylated tyrosine (p-Tyr) in unactivated platelets in the presence of secondary mediator inhibitors (n = 3). (B) s5-CD300a (40 nM, 200 nM for monomer) inhibits rhodocytin-induced (50 nM) platelet aggregation in humans and mice, which is abolished by anti-human/mouse CD148 antibody (10 µg/mL) (n ≥ 3). (C) s5-CD300a, not a control protein s5-CD200, inhibits Src and Lat phosphorylation by rhodocytin (n = 3). (D) s5-CD300a (40 nM, 200 nM for monomer) inhibits rhodocytin-induced (50 nM) platelet aggregation in human platelet-rich plasma. (E) s5-CD148 induces CD300a tyrosine phosphorylation in CD300a-transfected HEK293T cells. IP, immunoprecipitation; ′′, minutes.
The ability of CD300a to regulate platelet aggregation was tested against the hemi-TAM receptor CLEC-2. s5-CD300a had no effect on its own, but it inhibited rhodocytin-induced platelet aggregation (from 70.7% ± 3.5% to 18.2% ± 3.2%; Figure 2B) and tyrosine phosphorylation of Src family kinases and the adapter protein Lat (Figure 2C). The control pentamer s5-CD200 had no effect. Monomeric CD300a did not induce tyrosine phosphorylation or inhibit platelet activation by rhodocytin, providing evidence that its inhibitory action requires clustering of CD148 (data not shown). The effect of s5-CD300a was blocked by a polyclonal antibody to CD148, confirming that it is mediated by the interaction of the 2 proteins (Figure 2B). The polyclonal antibody had no effect on aggregation by itself (data not shown). s5-CD300a also inhibited rhodocytin-induced aggregation in mouse platelets (from 75.4% ± 4.9% to 45.1% ± 4.6%; Figure 2B). This suggests that the molecular mechanism is conserved across species. Furthermore, s5-CD300a does inhibit rhodocytin-induced platelet aggregation in the presence of plasma (from 85.1% ± 4.7% to 14.1% ± 3.2%; Figure 2D). In contrast, CD300a had no significant effect on the response to collagen (data not shown). This is intriguing because GPVI and CLEC-2 are ITAM receptors, and they share the common downstream signaling pathway. CD300a also had no effect on thrombin-induced platelet aggregation (supplemental Figure 4). In addition, we transfected full-length CD300a into HEK293T cells (which lack endogenous CD300a, as shown by flow cytometry; data not shown). s5-CD148 stimulation of the transfected cells led to CD300a tyrosine phosphorylation in a time-dependent manner (Figure 2E).
In summary, we identified several novel interactions between leukocyte and platelet proteins, including CD300a and platelet CD148, using a large-scale systematic screen for platelet-monocyte protein interactions. The binding of CD300a to CD148 inhibits CLEC-2–mediated platelet signaling and aggregation. We speculate that this novel interaction may play a role in monocyte/macrophage platelet interaction in inflammation and inflammatory-based disease.
Acknowledgments: The authors thank Gavin Wright for generously sharing part of the plasmid library used for this study.
This work was supported by a project grant from the British Heart Foundation (BHF) (PG/16/53/32242) and the Centre of Membrane Proteins and Receptors. S.P.W. is a BHF Chair (CH 003/03).
Contribution: R. Barroso performed experiments, analyzed the data, and wrote the manuscript; Y.S. designed the study, performed experiments, analyzed the data, and wrote the manuscript; and G.E.R., R. Bicknell, and S.P.W designed the study and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Yi Sun, Institute of Cardiovascular Sciences, College of Medical and Dental Sciences, 1st Floor IBR, University of Birmingham, Edgbaston, Birmingham B15 2TT, United Kingdom; e-mail: y.sun.3@bham.ac.uk.

