

Key Points
BET inhibitor RO6870810+venetoclax+rituximab has a manageable safety profile in relapsed/refractory diffuse large B-cell lymphoma.
A clinically relevant signal of antitumor activity serves as a proof of principle for this combination, warranting further study.

Abstract
Bromodomain and extraterminal (BET) proteins are transcriptional activators for multiple oncogenic processes in diffuse large B-cell lymphoma (DLBCL), including MYC, BCL2, E2F, and toll-like receptor signaling. We report results of a phase 1b dose-escalation study of the novel, subcutaneous BET inhibitor RO6870810 (RO) combined with the BCL-2 inhibitor venetoclax, and rituximab, in recurrent/refractory DLBCL. RO was delivered for 14 days of a 21-day cycle, whereas venetoclax was delivered continuously. A 3 + 3 escalation design was used to determine the safety of the RO+venetoclax doublet; rituximab was added in later cohorts. Thirty-nine patients were treated with a median of 2.8 cycles (range, 1-11). Dose-limiting toxicities included grade 3 febrile neutropenia, grade 4 diarrhea, and hypomagnesemia for the doublet; and grade 3 hyperbilirubinemia and grade 4 diarrhea when rituximab was added. The doublet maximum tolerated dose (MTD) was determined to be 0.65 mg/kg RO+600 mg venetoclax; for RO+venetoclax+rituximab, the MTDs were 0.45 mg/kg, 600 mg, and 375 mg/m2, respectively. The most frequent grade 3 and 4 adverse events were neutropenia (28%) and anemia and thrombocytopenia (23% each). Responses were seen in all cohorts and molecular subtypes. Sustained decreases in CD11b on monocytes indicated pharmacodynamic activity of RO. Overall response rate according to modified Lugano criteria was 38.5%; 48% of responses lasted for ≥180 days. Complete response was observed in 8 patients (20.5%). Optimization of the treatment schedule and a better understanding of predictors of response would be needed to support broader clinical use. This trial is registered on www.clinicaltrials.gov as NCT03255096.
Introduction
Bromodomain and extraterminal (BET) proteins have been implicated in malignant transformation and in resistance to anticancer therapy.1 Preclinical studies support the strategy of inhibiting the interaction between BET proteins and acetylated lysine to achieve antitumor effects. Direct-acting BET inhibitors use acetyl-lysine competitive binding to displace BET proteins from chromatin, resulting in preclinical activity in a variety of tumor models.2 The selectivity of these compounds for transformed cells arises from the localization of BET proteins to superenhancers that regulate cell-specific and oncogenic transcriptional programs, including those governed by the oncogene encoding the transcription factor MYC.1 In addition to MYC downregulation, BET inhibition has been shown to decrease oncogenic nuclear factor-B activity, decrease expression of ERK1/2, programmed death-ligand 1, CXCL12, and BCL2, thereby affecting immunity, cancer cell metastasis.3-8 and lymphoma proliferation.9 Patients with diffuse large B-cell lymphoma (DLBCL) that has relapsed or refractory (R/R) to first-line therapies or who are not candidates for high-dose therapy with autologous stem cell transplantation (ASCT), and those with high-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements (HGBL-DH/TH) represent a poor-prognosis group and a high unmet clinical need.10,11 Similarly, double-expressor DLBCLs (DE-DLBCLs), characterized by higher protein expression of MYC and BCL2, seem to be associated with lower rates of response and poorer survival after upfront R‐CHOP.12,13 Preclinical studies using BET inhibitor JQ1 in DLBCL models demonstrated potent, single agent in vivo activity. Clinical responses among heavily pretreated patients with relapsed DLBCL were observed in the phase 1 First in Man (FiM) monotherapy trial of the BET inhibitor RO6870810 (RO; NCT01987362).14 Partial responses (assessed by Lugano criteria15 ) were noted among patients with both MYC and MYC+BCL2 double-expression phenotype disease. BET protein inhibitors such as RO therefore may offer novel therapeutic options in DLBCL, double-expression lymphoma, and HGBL-DH/TH. However, compared with the administration as a single agent, rational combinations for BET inhibitors may improve clinical activity. Venetoclax, a small-molecule BCL2 inhibitor, has demonstrated single-agent responses in relapsed DLBCL, but these were short lived.16 The coadministration of BCL2 (venetoclax) and BET (JQ1) inhibitors produced the most potent synergistic effects among combinations tested in cell lines expressing MYC and bcl-2 proteins (DE-DLBCL cell lines) and primary patient-derived HGBL-DH models.17 Rituximab has been shown to improve the efficacy of venetoclax in the setting of chronic lymphocytic leukemia through sensitization of cells to apoptosis, and it does so with minimal additional toxicity. Nonoverlapping mechanisms of action and toxicities of RO, venetoclax, and rituximab suggest the possibility of enhanced activity when these agents are coadministered in the clinic. The demonstration of potent synergy with BET and BCL2 inhibitors provides an additional compelling rationale.17 We present a phase 1b study that explored RO given with venetoclax, with or without rituximab, in patients with R/R DLBCL and/or HGBL-DH/TH after standard first-line regimens.
Materials and methods
Study design and participants
Study NP39461 (NCT03255096) was an open-label, dose-escalation study with the objective of evaluating safety and dose-limiting toxicities (DLTs) and establishing the maximum tolerated doses (MTDs) or maximum dose administered of RO and venetoclax and the recommended doses of RO and venetoclax to be co-administered with rituximab. Patients received escalating doses of RO (0.3-0.65 mg/kg subcutaneously (SC) on days 1 to 14 of 21-day cycles) in a standard 3 + 3 design. RO was combined with venetoclax, which was administered orally starting at a dose of 400 mg once a day with subsequent investigation of 600 and 800 mg doses for additional cohorts, with RO (supplemental Figure 1). Patients were treated until disease progression, unacceptable toxicities or withdrawal from treatment for other reasons, or death.
Once the MTD/maximum dose administered was determined, additional patients were enrolled to receive the recommended doses of RO and venetoclax in combination with rituximab at a dose of 375 mg/m2 bovine serum albumin administered IV every week during the first cycle and on day 1 of all cycles thereafter.
Patients eligible for study inclusion had DLBCL and/or HGBL-DH/TH, R/R to first-line or subsequent therapies, who had received at least 1 prior chemotherapy regimen that included an anti-CD20 targeting agent, with no curative option. Patients were relapsed or refractory to ≥1 course of chemotherapy, including an anti-CD20 monoclonal antibody, and were not eligible for ASCT (including those with chemorefractory disease). Patients with transformed follicular lymphoma (FL) were eligible, provided DLBCL or HGBL-DH/TH histology was biopsy confirmed before study entry and a treatment regimen as described had been administered. All patients were ≥18 years of age, had an Eastern Cooperative Oncology Group (ECOG) performance status ≤1, and had acceptable organ function. Life expectancy was at least 3 months. This study was approved by local institutional review boards and conducted in accordance with the protocol, Good Clinical Practice standards, and the Declaration of Helsinki. All enrolled patients gave written informed consent.
Safety assessments
Patients were considered evaluable for safety if they had received ≥1 injection of study drug. All adverse events (AEs) were graded per the National Cancer Institute’s Common Terminology Criteria for Adverse Events, version 4.03.
DLTs were defined as AEs during cycle 1 that were at least possibly related to the study drug and met 1 of the following National Cancer Institute’s Common Terminology Criteria (version 4.03) criteria: grade 4 neutropenia lasting ≥5 days or grade 3 or 4 neutropenia with fever and/or infection; grade 4 thrombocytopenia (or grade 3 with bleeding); grade 4 anemia; grade 3 or 4 nonhematologic toxicity (excluding grade 3 vomiting and grade 3 diarrhea, including the clinical sequelae [eg, electrolyte abnormalities] occurring with suboptimal prophylactic and curative treatment with either toxicity and excluding alopecia); grade 3 or 4 skin ulceration or other skin and SC tissue disorders related to the SC injection of RO; or a dosing delay >14 days caused by treatment-emergent AEs or related severe laboratory abnormalities. Grade ≤3 drug-related fever, skin, or SC tissue disorders localized at the site of injection including grade ≤3 rash, pruritus, skin induration, or pain of skin were not considered DLTs.
The MTD was defined as the highest tested dose below the dose at which a DLT was observed in ≥2 patients.
Changes in vital signs, physical examination and ECG findings, clinical laboratory results, and the incidence of anti-drug antibodies to rituximab (during and after combination administration of RO+venetoclax and RO+venetoclax coadministered with rituximab) were also assessed.
Growth factors were prohibited within 7 days before enrollment but were allowed for treatment of cytopenias during the study.
Efficacy assessments
Evaluation of the anticancer activity of RO was a secondary objective, as measured by time to first and best response, overall response rate (ORR), progression-free survival (PFS), duration of response, and overall survival.
Response assessment in patients with DLBCL was made by Independent Radiological Centre Review and followed the Lugano classification.15 Radiological assessment consisted of diagnostic computed tomography (CT) at baseline and then every 2 cycles (standard of care) and combined positron emission tomography (PET)-CT at baseline and then every 4 cycles or to confirm complete remission (CR).
Patients who received ≥1 injection of study drug, had ≥1 post-baseline tumor assessment per Lugano classification criteria, and had no major protocol deviations were considered evaluable for efficacy. Responders were defined as patients who achieved a confirmed CR or partial response (PR). PFS was defined as the time from the date of first study drug administration to the first date of objectively determined progressive disease or death from any cause. PFS was censored at the date of the most recent objective progression-free observation for patients who were still alive at the time of analysis and without evidence of tumor progression. For patients who received subsequent anticancer therapy before objective disease progression or death, PFS was censored at the date of the last objective progression-free observation before the date of subsequent therapy.
Pharmacokinetic parameters
Blood samples were collected to evaluate concentrations of RO and were analyzed by validated methods. Multiple samples were collected for assessment of RO on days 1 and 15 of cycle 1, and day 1 of subsequent even-numbered cycles.
Pharmacodynamics and biomarkers
For the pharmacodynamic assessments, venous blood samples were obtained weekly during cycle 1. The absolute number of CD11b+ cells within the CD14+ positive monocyte population was assessed as a surrogate pharmacodynamic marker by flow cytometry, before and after the first administration of RO.
Statistical methods
Formal hypothesis testing was not performed for this phase 1b study. The sample size was based on a standard 3 + 3 dose-escalation design and was considered sufficient to evaluate the safety and clinical activity of the combinations. AEs were coded using the current Medical Dictionary for Regulatory Activities, and safety and clinical efficacy were summarized by descriptive statistics. The 95% confidence interval (CI) for the objective response rate was calculated using the Wilson’s score method with continuity correction, and PFS was analyzed using the Kaplan-Meier method. For the pharmacokinetic analyses, key exposure parameters such as maximum plasma concentration and area under the curve were summarized with descriptive statistics. The study design with the different dose cohorts is shown in Figure 1.

Concentration-time profiles for RO in combination with venetoclax across the dose range 0.3 to 0.65 mg/kg.
Concentration-time profiles for RO in combination with venetoclax across the dose range 0.3 to 0.65 mg/kg.
All authors had a role in analyzing the data and had access to the primary clinical trial data.
Results
Patient demographics and disposition
Thirty-nine patients with R/R DLBCL were recruited into the study, and those who received at least 1 dose of study drug were evaluable for safety and efficacy analysis (Table 1).
Baseline disease characteristics
| Baseline characteristics | Population (N = 39) |
|---|---|
| Disease stage IV, n (%) | 30/37 (81.1) |
| Transformed FL, n (%) | 8/38 (21.5) |
| International Prognostic Index score, median (range) (n = 36) | 3 (0-4) |
| Prior DLBCL treatments, mean (min-max) | 3.3 (1-6) |
| CAR T, n (%) | 2 (5.1) |
| Bispecific antibodies, n (%) | 5 (12.8) |
| Interval between prior treatment and study treatment | |
| ≤1 mo | 11 (28.2) |
| >1 mo and ≤ 3 mo | 17 (43.6) |
| > 3 mo | 11 (28.2) |
| Cell of origin, n (%) | |
| GCB | 24/33 (72.7) |
| ABC | 7/33 (21.2) |
| MYC+BCL2 DE-DLBCL, n (%) | 10/18 (55.5) |
| MYC translocation, n (%) | 1/30 (3.3) |
| BCL2 translocation, n (%) | 9/30 (30.0) |
| Baseline characteristics | Population (N = 39) |
|---|---|
| Disease stage IV, n (%) | 30/37 (81.1) |
| Transformed FL, n (%) | 8/38 (21.5) |
| International Prognostic Index score, median (range) (n = 36) | 3 (0-4) |
| Prior DLBCL treatments, mean (min-max) | 3.3 (1-6) |
| CAR T, n (%) | 2 (5.1) |
| Bispecific antibodies, n (%) | 5 (12.8) |
| Interval between prior treatment and study treatment | |
| ≤1 mo | 11 (28.2) |
| >1 mo and ≤ 3 mo | 17 (43.6) |
| > 3 mo | 11 (28.2) |
| Cell of origin, n (%) | |
| GCB | 24/33 (72.7) |
| ABC | 7/33 (21.2) |
| MYC+BCL2 DE-DLBCL, n (%) | 10/18 (55.5) |
| MYC translocation, n (%) | 1/30 (3.3) |
| BCL2 translocation, n (%) | 9/30 (30.0) |
Overall, the median age of patients was 65 years (range, 28-81 years). Nineteen patients (48.7%) were male and 20 (51.3%) were female. The majority of patients were White (37 patients [94.9%]). Of the 39 enrolled patients, 19 patients (48.7%) had an Eastern Cooperative Oncology Group (ECOG) score of 1, 18 patients (46.2%) had an ECOG score of 0, and the remaining 2 patients (5.1%) had an ECOG score of 2. Baseline disease characteristics are presented in Table 1. Of note, patients had a mean of 3.3 prior DLBCL treatments. Twenty-nine patients (74%) were refractory to the immediate prior therapy, and 72% of patients received the study treatment in <3 months after the end of the prior therapy. All 39 patients discontinued the treatment by the time of data cutoff on 7 November 2019; 10 patients (25.6%) reported AEs that led to treatment discontinuation; 24 patients (61.5%) recorded progressive disease, symptomatic deterioration, or lack of efficacy as the primary reasons for discontinuing treatment; 1 patient (2.6%) withdrew from treatment; and 4 patients (10.3%) discontinued study treatment because of other or unknown reasons. Patients completed a median of 2.8 treatment cycles (range, 1-11).
Safety
All 39 enrolled patients (100%) experienced at least 1 treatment-emergent AE.
The majority of patients experienced treatment-emergent grade ≥3 AEs (34 patients [87.2%]), or serious AEs (SAEs; 25 patients [64.1%]). Of those, 23 patients (59%) had related grade ≥3 AEs, and 11 (28.2%) had related SAEs (supplemental Table 1).
Five patients experienced 5 DLTs. DLTs occurred in cohort 3 (grade 3 febrile pneumonia; RO 0.45 mg/kg+venetoclax 600 mg), cohort 4 (grade 4 hypomagnesemia, grade 4 diarrhea; RO 0.45 mg/kg+venetoclax 800 mg), and cohort 5A (grade 3 hyperbilirubinemia, grade 4 diarrhea; RO 0.65 mg/kg+venetoclax 600 mg+rituximab 375 mg/m2). All DLTs were considered serious except for the DLT of hypomagnesemia (supplemental Table 1). The doublet MTD was determined to be 0.65 mg/kg RO+600 mg venetoclax, and the triplet MTD was 0.45 mg/kg RO+600 mg venetoclax+375 mg/m2 rituximab.
The most common AEs (experienced by ≥30% patients) were injection site reaction (24 patients [61.5%]), diarrhea (23 patients [59.0%]), anemia and nausea (18 patients [46.2%] each), thrombocytopenia (13 patients [33.3%]), and neutropenia (12 patients [30.8%]). AEs experienced by ≥15% of the patients are summarized in Table 2.
Treatment-emergent AEs by cohort, all grades, in ≥5% of patients
| Cohort 1 | Cohort 2 | Cohort 3 | Cohort 3A | Cohort 4 | Cohort 5 | Cohort 5A | Total | |
|---|---|---|---|---|---|---|---|---|
| RO 0.30 mg/kg + venetoclax 400 mg, n (%) | RO 0.45 mg/kg + venetoclax 400 mg, n (%) | RO 0.45 mg/kg + venetoclax 600 mg, n (%) | RO 0.45 mg/kg + venetoclax 600 mg + rituximab 375 mg/m2, n (%) | RO 0.45 mg/kg + venetoclax 800 mg, n (%) | RO 0.65 mg/kg + venetoclax 600 mg, n (%) | RO 0.65 mg/kg + venetoclax 600 mg + rituximab 375 mg/m2, n (%) | ||
| Patients, n | 3 | 6 | 9 | 7 | 4 | 4 | 6 | 39 |
| Injection site reaction | 2 (66.7) | 6 (100) | 6 (66.7) | 3 (42.9) | 2 (50.0) | 2 (50.0) | 3 (50.0) | 24 (61.5) |
| Diarrhea | 0 | 4 (66.7) | 5 (55.6) | 3 (42.9) | 3 (75.0) | 4 (100) | 4 (66.7) | 23 (59.0) |
| Nausea | 1 (33.3) | 4 (66.7) | 5 (55.6) | 3 (42.9) | 2 (50.0) | 2 (50.0) | 1 (16.7) | 18 (46.2) |
| Anemia | 0 | 4 (66.7) | 4 (44.4) | 1 (14.3) | 2 (50.0) | 3 (75.0) | 4 (66.7) | 18 (46.2) |
| Thrombocytopenia | 0 | 2 (33.3) | 4 (44.4) | 2 (28.6) | 0 | 3 (75.0) | 2 (33.3) | 13 (33.3) |
| Neutropenia | 1 (33.3) | 1 (16.7) | 5 (55.6) | 2 (28.6) | 0 | 2 (50.0) | 1 (16.7) | 12 (30.8) |
| Fatigue | 0 | 2 (33.3) | 2 (22.2) | 1 (14.3) | 2 (50.0) | 2 (50.0) | 2 (33.3) | 11 (28.2) |
| Hypomagnesemia | 0 | 2 (33.3) | 2 (22.2) | 0 | 2 (50.0) | 2 (50.0) | 2 (33.3) | 10 (25.6) |
| Decreased appetite | 1 (33.3) | 1 (16.7) | 1 (11.1) | 0 | 2 (50.0) | 1 (25.0) | 2 (33.3) | 8 (20.5) |
| Hypokalemia | 0 | 0 | 1 (11.1) | 1 (14.3) | 1 (25.0) | 1 (25.0) | 3 (50.0) | 7 (17.9) |
| Vomiting | 0 | 3 (50.0) | 2 (22.2) | 0 | 1 (25.0) | 1 (25.0) | 0 | 7 (17.9) |
| Peripheral edema | 0 | 1 (16.7) | 2 (22.2) | 2 (28.6) | 0 | 0 | 1 (16.7) | 6 (15.4) |
| Pyrexia | 0 | 1 (16.7) | 2 (22.2) | 1 (14.3) | 2 (50.0) | 0 | 0 | 6 (15.4) |
| Herpes zoster | 0 | 0 | 2 (22.2) | 0 | 2 (50.0) | 1 (25.0) | 1 (16.7) | 6 (15.4) |
| Cohort 1 | Cohort 2 | Cohort 3 | Cohort 3A | Cohort 4 | Cohort 5 | Cohort 5A | Total | |
|---|---|---|---|---|---|---|---|---|
| RO 0.30 mg/kg + venetoclax 400 mg, n (%) | RO 0.45 mg/kg + venetoclax 400 mg, n (%) | RO 0.45 mg/kg + venetoclax 600 mg, n (%) | RO 0.45 mg/kg + venetoclax 600 mg + rituximab 375 mg/m2, n (%) | RO 0.45 mg/kg + venetoclax 800 mg, n (%) | RO 0.65 mg/kg + venetoclax 600 mg, n (%) | RO 0.65 mg/kg + venetoclax 600 mg + rituximab 375 mg/m2, n (%) | ||
| Patients, n | 3 | 6 | 9 | 7 | 4 | 4 | 6 | 39 |
| Injection site reaction | 2 (66.7) | 6 (100) | 6 (66.7) | 3 (42.9) | 2 (50.0) | 2 (50.0) | 3 (50.0) | 24 (61.5) |
| Diarrhea | 0 | 4 (66.7) | 5 (55.6) | 3 (42.9) | 3 (75.0) | 4 (100) | 4 (66.7) | 23 (59.0) |
| Nausea | 1 (33.3) | 4 (66.7) | 5 (55.6) | 3 (42.9) | 2 (50.0) | 2 (50.0) | 1 (16.7) | 18 (46.2) |
| Anemia | 0 | 4 (66.7) | 4 (44.4) | 1 (14.3) | 2 (50.0) | 3 (75.0) | 4 (66.7) | 18 (46.2) |
| Thrombocytopenia | 0 | 2 (33.3) | 4 (44.4) | 2 (28.6) | 0 | 3 (75.0) | 2 (33.3) | 13 (33.3) |
| Neutropenia | 1 (33.3) | 1 (16.7) | 5 (55.6) | 2 (28.6) | 0 | 2 (50.0) | 1 (16.7) | 12 (30.8) |
| Fatigue | 0 | 2 (33.3) | 2 (22.2) | 1 (14.3) | 2 (50.0) | 2 (50.0) | 2 (33.3) | 11 (28.2) |
| Hypomagnesemia | 0 | 2 (33.3) | 2 (22.2) | 0 | 2 (50.0) | 2 (50.0) | 2 (33.3) | 10 (25.6) |
| Decreased appetite | 1 (33.3) | 1 (16.7) | 1 (11.1) | 0 | 2 (50.0) | 1 (25.0) | 2 (33.3) | 8 (20.5) |
| Hypokalemia | 0 | 0 | 1 (11.1) | 1 (14.3) | 1 (25.0) | 1 (25.0) | 3 (50.0) | 7 (17.9) |
| Vomiting | 0 | 3 (50.0) | 2 (22.2) | 0 | 1 (25.0) | 1 (25.0) | 0 | 7 (17.9) |
| Peripheral edema | 0 | 1 (16.7) | 2 (22.2) | 2 (28.6) | 0 | 0 | 1 (16.7) | 6 (15.4) |
| Pyrexia | 0 | 1 (16.7) | 2 (22.2) | 1 (14.3) | 2 (50.0) | 0 | 0 | 6 (15.4) |
| Herpes zoster | 0 | 0 | 2 (22.2) | 0 | 2 (50.0) | 1 (25.0) | 1 (16.7) | 6 (15.4) |
The grade ≥3 AEs experienced by ≥10% of patients were neutropenia (11 patients [28.2%]); anemia and thrombocytopenia (9 patients [23.1%]); hypokalemia (5 patients [12.8%]); and febrile neutropenia (4 patients [10.3%]; Table 3).
Treatment-emergent AEs by cohort, grades 3 and 4, in ≥10% of patients
| Cohort 1 | Cohort 2 | Cohort 3 | Cohort 3A | Cohort 4 | Cohort 5 | Cohort 5A | Total | |
|---|---|---|---|---|---|---|---|---|
| RO 0.30 mg/kg + Venetoclax 400 mg, n (%) | RO 0.45 mg/kg + Venetoclax 400 mg, n (%) | RO 0.45 mg/kg + Venetoclax 600 mg, n (%) | RO 0.45 mg/kg + Venetoclax 600 mg + Rituximab 375 mg/m2, n (%) | RO 0.45 mg/kg + Venetoclax 800 mg, n (%) | RO 0.65 mg/kg + Venetoclax 600 mg, n (%) | RO 0.65 mg/kg + Venetoclax 600 mg + Rituximab 375 mg/m2, n (%) | ||
| Patients, n | 3 | 6 | 9 | 7 | 4 | 4 | 6 | 39 |
| Neutropenia | 1 (33.3) | 1 (16.7) | 5 (55.6) | 1 (14.3) | 0 | 2 (50.0) | 1 (16.7) | 11 (28.2) |
| Anemia | 0 | 3 (50.0) | 1 (11.1) | 1 (14.3) | 1 (25.0) | 2 (50.0) | 1 (16.7) | 9 (23.1) |
| Thrombocytopenia | 0 | 2 (33.3) | 2 (22.2) | 2 (28.6) | 0 | 2 (50.0) | 1 (16.7) | 9 (23.1) |
| Hypokalemia | 0 | 0 | 0 | 1 (14.3) | 1 (25.0) | 0 | 3 (50.0) | 5 (12.8) |
| Febrile Neutropenia | 0 | 1 (16.7) | 2 (22.2) | 0 | 0 | 0 | 1 (16.7) | 4 (10.3) |
| Cohort 1 | Cohort 2 | Cohort 3 | Cohort 3A | Cohort 4 | Cohort 5 | Cohort 5A | Total | |
|---|---|---|---|---|---|---|---|---|
| RO 0.30 mg/kg + Venetoclax 400 mg, n (%) | RO 0.45 mg/kg + Venetoclax 400 mg, n (%) | RO 0.45 mg/kg + Venetoclax 600 mg, n (%) | RO 0.45 mg/kg + Venetoclax 600 mg + Rituximab 375 mg/m2, n (%) | RO 0.45 mg/kg + Venetoclax 800 mg, n (%) | RO 0.65 mg/kg + Venetoclax 600 mg, n (%) | RO 0.65 mg/kg + Venetoclax 600 mg + Rituximab 375 mg/m2, n (%) | ||
| Patients, n | 3 | 6 | 9 | 7 | 4 | 4 | 6 | 39 |
| Neutropenia | 1 (33.3) | 1 (16.7) | 5 (55.6) | 1 (14.3) | 0 | 2 (50.0) | 1 (16.7) | 11 (28.2) |
| Anemia | 0 | 3 (50.0) | 1 (11.1) | 1 (14.3) | 1 (25.0) | 2 (50.0) | 1 (16.7) | 9 (23.1) |
| Thrombocytopenia | 0 | 2 (33.3) | 2 (22.2) | 2 (28.6) | 0 | 2 (50.0) | 1 (16.7) | 9 (23.1) |
| Hypokalemia | 0 | 0 | 0 | 1 (14.3) | 1 (25.0) | 0 | 3 (50.0) | 5 (12.8) |
| Febrile Neutropenia | 0 | 1 (16.7) | 2 (22.2) | 0 | 0 | 0 | 1 (16.7) | 4 (10.3) |
Twelve (31%) patients received pegfilgrastim/filgrastim, 13 (33%) received blood transfusion, and 7 received (18%) platelets; 5 (13%) patients received both.
The SAEs experienced by ≥5% of patients were pneumonia (4 patients [10.3%]), febrile neutropenia (3 patients [7.7%]), and diarrhea and pyrexia (2 patients [5.1%] in each PT). Eleven patients (28.2%) experienced 17 SAEs considered related to the study treatment by the investigator. The related SAEs experienced in >1 patient (≥5%) were pneumonia and diarrhea (2 patients [5.1%] in each PT).
AEs leading to treatment discontinuation
Ten patients (25.6%) had a total of 10 AEs leading to discontinuation of the study treatment, with pneumonia (2 patients [5.1%]) being reported in >1 patient (≥5%; supplemental Table 1). The remaining of these AEs were reported in 1 patient each: hepatitis, hyperbilirubinemia, thrombocytopenia, gastric hemorrhage, craniocerebral injury, hypomagnesaemia, anxiety, and pulmonary embolism. In 4 patients (10.2%), AEs leading to treatment discontinuation were considered to be related to the study treatment by the investigator.
Twenty-one deaths (53.8%) were reported in the study: 18 deaths (46.2%) due to progressive disease, 1 death due to AE (pneumonia, considered unrelated to study drug by the investigator), 1 due to disease relapse, and 1 due to an infusion reaction to a subsequent treatment during the long-term follow-up period. Of the 21 deaths, 14 occurred in the long-term follow-up period, and 7 occurred in the period before long-term follow-up.
Pharmacokinetics
Plasma concentration-time profiles for RO administered in combination with venetoclax or in combination with venetoclax and rituximab are presented in Figure 1. RO demonstrated rapid absorption across dose levels. Maximum concentration was generally achieved within 1 hour (median, 0.5 hour).
Pharmacodynamics
Chromatin immunoprecipitation sequencing previously identified a strong BRD4-driven superenhancer near the CD11b promoter, and displacement of bound BRD4 from the superenhancer element by a BET inhibitor resulted in diminished CD11b gene expression. In this study, CD11b expression on peripheral blood mononuclear cells was measured by flow cytometry before the dose and at various time points between days 1 and 15 of cycle 1. Treatment with RO led to sustained decreases in CD11b during the dosing period. At day 15, the CD11b cell counts were reduced to a median of 42% of baseline counts (interquartile range, 34% to 67%) (Figure 2). There was no difference in CD11b reduction according to response (P = .94), but some evidence that the 7 responders with a >50% reduction in CD11b counts had a longer response (median, 127 days) compared with the 5 responders with a <50% reduction (median, 43 days), but this finding is based on very limited data.
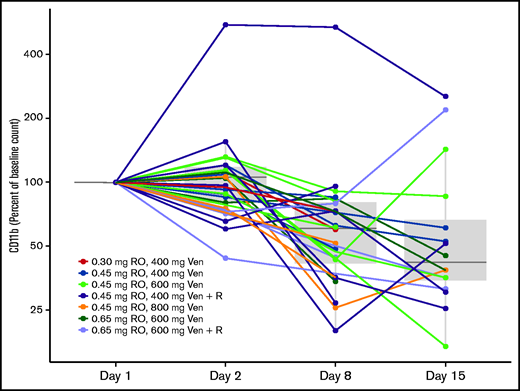
RO pharmacodynamic profiling as evidence of target engagement. CD11b (molecules of equivalent soluble fluorochrome) change from baseline (%) over time. Ven, venetoclax; R, rituximab.
RO pharmacodynamic profiling as evidence of target engagement. CD11b (molecules of equivalent soluble fluorochrome) change from baseline (%) over time. Ven, venetoclax; R, rituximab.
Clinical activity
All 39 patients were included in the analysis of clinical activity. Response assessments by the investigator were available in 33 patients. The ORR according to modified Lugano criteria was 38.5%. Complete response was observed in 8 patients (20.5%), and PR was observed in 7 patients (17.9%). Six patients (15.4%) had stable disease, and 12 (30.8%) had progressive disease. In 6 patients (15.4%) response information was missing as those patients withdrew during cycle 1. Reasons were symptomatic deterioration, AEs and clinical progression (2 patients each). For response information according to cohort, see supplemental Table 2. The percentage change from baseline of the sum of longest diameters of the tumor lesions over time is shown in Figure 3. Type and duration of response is shown in Figure 4. Seventy-seven percent (95% CI, 44-92) of responses lasted for ≥90 days and 48% (95% CI, 4-76) lasted for ≥180 days. The majority of the 8 complete responders (83%) had a duration of response of ≥90 days.
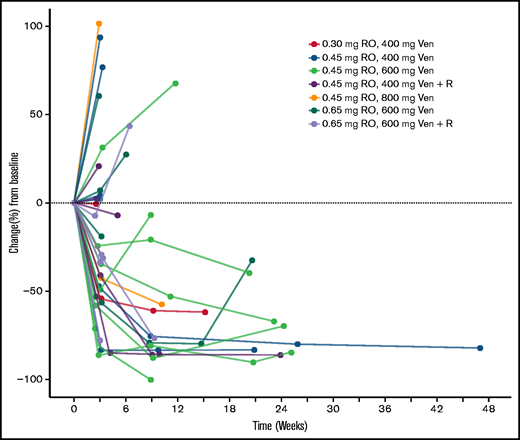
Sum of longest diameters over time. Percentage change from baseline. Ven, venetoclax; R, rituximab.
Sum of longest diameters over time. Percentage change from baseline. Ven, venetoclax; R, rituximab.
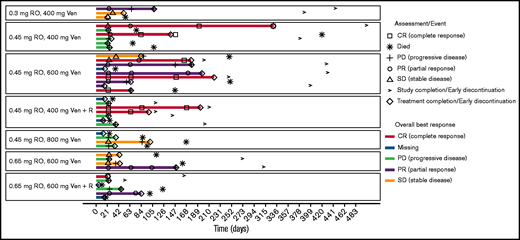
Swimmers plot according to cohort. Timeline from the first date of treatment to the end of treatment. If the end of treatment was not reached, then the end was last visit, last laboratory assessment, or last AE.
Swimmers plot according to cohort. Timeline from the first date of treatment to the end of treatment. If the end of treatment was not reached, then the end was last visit, last laboratory assessment, or last AE.
The ORR and CR rate were similar between cell-of-origin (COO) subtypes. A high response rate was detected in transformed FL (ORR, 62%; Table 4).
Response in total population, in transformed FL and by COO subtype
| GCB | ABC | Unclassified | Total | |
|---|---|---|---|---|
| Patients, n | 15 | 5 | 19 | 39 |
| ORR, n (%) | 7 (46.7) | 1 (20.0) | 7 (36.8) | 15 (38.5) |
| CR, n (%) | 3 (20.0) | 0 | 5 (26.3) | 8 (20.5) |
| N-transformed FL | ||||
| Patients, n | 3 | 0 | 5 | 8 |
| ORR-transformed FL, n (%) | 2 (67.7) | 0 | 3 (60.0) | 5 (62.5) |
| GCB | ABC | Unclassified | Total | |
|---|---|---|---|---|
| Patients, n | 15 | 5 | 19 | 39 |
| ORR, n (%) | 7 (46.7) | 1 (20.0) | 7 (36.8) | 15 (38.5) |
| CR, n (%) | 3 (20.0) | 0 | 5 (26.3) | 8 (20.5) |
| N-transformed FL | ||||
| Patients, n | 3 | 0 | 5 | 8 |
| ORR-transformed FL, n (%) | 2 (67.7) | 0 | 3 (60.0) | 5 (62.5) |
Ten of 18 patients (55.6%) for whom the MYC and BCL2 baseline expression status was known were patients with DE-DLBCL. No patient had double-hit lymphoma. One patient achieved a CR (ie, ORR in DE-DLBCL patients was 10%; Figure 5).
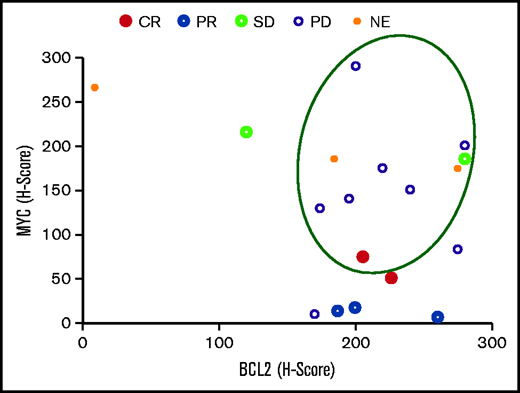
Clinical responses according to MYC and BCL2 expression. The circled events occurred in patients with DE-DLBCL. SD, stable disease; NE, not evaluable.
Clinical responses according to MYC and BCL2 expression. The circled events occurred in patients with DE-DLBCL. SD, stable disease; NE, not evaluable.
Discussion
RO+venetoclax+rituximab had a clinically relevant signal of activity that serves as a proof of principle of the rationale driving this trial. This was a highly refractory population, and some patients enjoyed a protracted response to therapy out of keeping with the monotherapy trials of venetoclax or RO. The combination was deliverable, but with some toxicities that would have to be addressed before further development of the combination. The key toxicities seen in this study were largely predictable based on what is known about RO and other BET inhibitors. They are qualitatively similar to those reported in the FiM trial for RO14 and those known for orally administered birabresib18 and molibresib.19 They include fatigue, anemia, and thrombocytopenia, and gastrointestinal side effects, including diarrhea and nausea. The SC administration of RO did not prevent gastrointestinal toxicities in this study, suggesting that these AEs may represent on-target class effects.
In the FiM trial of RO, grade 3 and 4 anemia and thrombocytopenia occurred in 8.1% and 9.5% of patients, respectively.14 In a phase 1 trial of venetoclax in patients with non-Hodgkin lymphoma, grade 3 and 4 anemia and thrombocytopenia occurred in 15% and 9% of patients, respectively.16 The higher incidence of these AEs in the trial reported herein indicates an additive effect for the BET inhibitor+venetoclax combination, relative to single-agent AE profiles. At the recommended dose for the triplet regimen, none of the hematologic toxicities led to treatment discontinuation. This was enabled by treatment interruptions for grade 3 and 4 hematologic events of up to 3 weeks and treatment continuation at lower dose levels if AEs improved to grade ≤2.
A lower dose of venetoclax (eg, 400 mg instead of 600 mg), combined with 0.65 mg of RO, may have resulted in a better safety profile. With a recommended single-agent dose in DLBCL of 1200 mg daily,16 venetoclax doses below 600 mg were not considered to be efficacious in DLBCL at the time of trial setup.
Decreases in CD11b in peripheral blood mononuclear cells were observed with RO treatment, supporting the putative mechanism of action that RO prevents BET coactivator loading at super-enhancers and are indicative of successful target engagement. For future clinical studies, an assessment beyond cycle 1 and a paired analysis of tumor tissue with peripheral blood samples may be instructive about the durability of this effect.
RO doublet and triplet combinations were active, with responses observed in patients with R/R DLBCL and transformed FL. The ORR of 38.5% and the CR rate of 20.5% across dose and combination cohorts compares favorably to responses observed with RO and venetoclax administered as a monotherapy. The observation that 48% of responders appear to have a duration of response of ≥180 days is an interesting signal in this heavily treated population. By contrast, the FiM trial of RO monotherapy enrolled 13 response-evaluable patients with DLBCL, of whom 2 achieved a PR.14 For venetoclax, Davids et al reported an ORR of 18% and a CR rate of 12% in 34 patients with DLBCL, with a median PFS of 1 month.16
Although the overall sample size was small, there is no indication that clinical activity of the combination treatment was higher in the subgroup of patients with DE-DLBCL. None of the trial patients was confirmed to have double-hit lymphoma, and further exploration of that subgroup is not supported by the efficacy data.
The high response rate in transformed FL is of interest, but requires further evaluation in a larger patient sample.
When the different cohorts were compared, there was no indication of a dose-response relationship and no added benefit of rituximab. Treatment duration, as indicated in the swimmers plot (Figure 4), shows no correlation to dose or added rituximab treatment. The response rate in the germinal center B-cell and activated B-cell subtypes appeared to be similar.
Pharmacokinetic analyses confirmed results from the FiM trial and showed rapid absorption, linearity for exposure across the dose range tested, and a half-life of >10 hour, supporting a once-daily SC dose.14 Importantly, exposure appears similar to that in previous studies, suggesting no impact of venetoclax on RO pharmacokinetics.
Although the BET inhibitor/venetoclax/rituximab combination resulted in higher response rates compared with that observed with single agents in patients with heavily pretreated R/R DLBCL, a synergistic effect of that treatment was not shown. Additional studies to understand markers of response and on-treatment pharmacodynamic effects would be needed to optimize this regimen for broader clinical use, but the priority of such studies seems to be low in light of other treatment options, such as anti-CD19+lenalidomide, T-cell–engaging bispecific antibodies, antibody-drug conjugates, and CAR-T cells, which have already been shown to provide clinical benefit.20
Acknowledgments
The authors thank the patients, their families, and the study teams at the participating centers. Medical Writers Fiona Russel-Yarde (FRY Medical Communications Ltd.) and Christian Seitz provided editorial assistance supported by funding from F. Hoffmann-La Roche Ltd.
This study was sponsored and funded by F. Hoffmann-La Roche Ltd.
Authorship
Contribution: M.D., M.D.M., D.R., and M.H. designed the clinical study; M.D., J.B., A.F.H., E.G.B., N.G., R.C., S.C.R., and M.H. collected data; E.N. performed statistical analysis; and all authors analyzed and interpreted the data and wrote, critically reviewed, and approved the manuscript for submission.
Conflict-of-interest disclosure: M.D. reported honoraria from Roche, Amgen, MSD, Janssen, BMS, Novartis; consultancy for Novartis, Roche, BMS, Gilead, and Janssen; participation on the speakers’ bureau for Novartis; and institutional research funding from Novartis, Roche, Takeda, Celgene, and MDS. J.B. reported honoraria from Roche, Takeda, Celgene, Novartis, and Gilead; consultancy for Takeda, Janssen, Celgene, and Gilead/Kyte; research funding from Celgene and Roche; and travel grants from Roche, Takeda, Celgene, Janssen, and Gilead. A.F.H. reported research funding from BMS, Genentech/Roche, Immune Design, Merck, Pharmacyclics, and Seattle Genetics; consultancy for Bristol-Myers Squibb, Genentech/Roche, Merck, Seattle Genetics, and Karyopharm; and travel grants from Bristol-Myers Squibb. E.G.B. reported consultancy for Janssen, AbbVie, Celgene, Gilead, Kiowa, EUSApharma; participation on the speakers’ bureau for Janssen, AbbVie, and Takeda; and travel grants from Janssen, AbbVie, and Roche. N.G. reported consultancy for Bristol Myers Squibb, TG Therapeutics, Seattle Genetics, Janssen, and Pharmacyclics; participation on the speakers’ bureau for Bristol Myers Squibb, Seattle Genetics, Janssen, Pharmacyclics, and AbbVie; and research funding from Bristol Myers Squibb, TG Therapeutics, Pharmacyclics, and Genentech. R.C. reports participation on speakers’ bureaus, consultancy, and travel funding from Janssen. S.C.R. reported consultancy for AstraZeneca, Celgene, Heron, Juno, Janssen, Karyopharm, Seattle Genetics, Verastem, Dova, and Kite; and research funding from Roche/Genentech. E.B., E.L.-T., J.B.-V., B.B., and M.D.M. were employees of Roche during the conduct of the trial. I.F., E.C., K.L., E.N., D.R., and M.K. are employees of Roche and own stock. M.H. reported research funding from and consultancy for Takeda, Roche, and Celgene.
Correspondence: Michael Dickinson, Clinical Haematology, Peter MacCallum Cancer Centre and Royal Melbourne Hospital, Locked Bag 1 A’Beckett St, Melbourne, VIC 8006, Australia; e-mail: michael.dickinson@petermac.org.

