

Key Points
Platelet interaction with polymerizing fibrin is avidity driven and requires activated αIIbβ3 but not fibrin cross-linking.
The mechanism by which αIIbβ3 interacts with polymerizing fibrin differs subtly from the interaction of αIIbβ3 with fibrinogen.

Abstract
The molecular basis of platelet-fibrin interactions remains poorly understood despite the predominance of fibrin in thrombi. We have studied the interaction of platelets with polymerizing fibrin by adding thrombin to washed platelets in the presence of the peptide RGDW, which inhibits the initial platelet aggregation mediated by fibrinogen binding to αIIbβ3 but leaves intact a delayed increase in light transmission (delayed wave; DW) as platelets interact with the polymerizing fibrin. The DW was absent in platelets from a patient with Glanzmann thrombasthenia, indicating a requirement for αIIbβ3. The DW required αIIbb3 activation and it was inhibited by the αIIbβ3 antagonists eptifibatide and the monoclonal antibody (mAb) 7E3, but only at much higher concentrations than needed to inhibit platelet aggregation initiated by a thrombin receptor activating peptide (T6). Surface plasmon resonance and scanning electron microscopy studies both supported fibrin having greater avidity for αIIbβ3 than fibrinogen rather than greater affinity, consistent with fibrin’s multivalency. mAb 10E5, a potent inhibitor of T6-induced platelet aggregation, did not inhibit the DW, suggesting that fibrin differs from fibrinogen in its mechanism of binding. Inhibition of factor XIII–mediated fibrin cross-linking by >95% reduced the DW by only 32%. Clot retraction showed a pattern of inhibition similar to that of the DW. We conclude that activated αIIbβ3 is the primary mediator of platelet-fibrin interactions leading to clot retraction, and that the interaction is avidity driven, does not require fibrin cross-linking, and is mediated by a mechanism that differs subtly from that of the interaction of αIIbβ3 with fibrinogen.
Introduction
Whereas vascular injury leads to sufficient thrombin (Thr) generation within 20 seconds to produce fibrin deposition,1,2 and whereas Thr also rapidly activates factor XIII, resulting in fibrin cross-linking via reciprocal transamidation of the C-terminal γ-chain peptides from adjacent fibrinogen molecules, platelets are likely to primarily encounter polymerizing fibrin and cross-linked fibrin in developing and mature thrombi.3-6 Moreover, the interactions of platelets with these fibrin species are also likely to contribute to clot retraction, which stabilizes clots and diminishes their susceptibility to fibrinolysis.7-10 Because patients with Glanzmann thrombasthenia, who lack or have an abnormal αIIbβ3 receptor, have diminished or absent clot retraction, it is likely that αIIbβ3 is required for fibrin interaction and clot retraction.11-13
Investigators have, however, reported that platelet interactions with fibrin can be supported by GPVI (reviewed by Slater et al,14 and Perrella et al,15 and Xu et al16 ) and GPIb, either directly or through interactions with von Willebrand factor,17-23 and thus, these receptors may also participate in the interaction of platelets with polymerizing fibrin and clot retraction.
The interaction between the αIIbβ3 Arg-Gly-Asp (RGD)-binding pocket, formed jointly by the αIIb β-propeller and β3 β-I domains, and the fibrinogen γ-chain C-terminal sequence 404 to 411, has been defined at the atomic level,24,25 but the sites on αIIbβ3 involved in fibrin binding may be different from those that mediate fibrinogen binding because: (1) γ-chain Lys406, which interacts with αIIb Asp224,25 is cross-linked to γ-Gln398/Gln399 in cross-linked fibrin,26,27 eliminating its ability to make an ionic interaction with Asp224; (2) other sites on fibrinogen and αIIbβ3, including sites exposed when fibrinogen is converted into fibrin,28,29 have been suggested to support interactions between fibrin and αIIbβ330,31 ; (3) monomeric fibrin made from γ′/γ′-fibrinogen, which lacks the γ-12 sequence, can interact with purified αIIbβ331 ; (4) treating αIIbβ3 with EDTA under conditions that lead to irreversible loss of the ability to bind fibrinogen does not inhibit clot retraction32,33 ; (5) in contrast to the ability of unactivated αIIbβ3 to support adhesion to immobilized, intact fibrinogen,34 our recent data indicate that platelet αIIbβ3 activation is required to support adhesion to D-dimer, a plasmin degradation product of cross-linked fibrin that contains the cross-link site6 ; (6) platelet interactions with fibrin also differ from those with fibrinogen with regard to ligand valency. Thus, polymerized fibrin’s marked multivalency may contribute to greater avidity, even with the same receptor-ligand affinity.35,36
To study the molecular basis of the interaction of platelets with polymerizing, cross-linked fibrin as well as the impact of fibrin’s multivalency, we developed an assay to separate the αIIbβ3-mediated interactions with fibrinogen from those with polymerizing fibrin. We also used surface plasmon resonance (SPR) to compare the affinity of monomolecular αIIbβ3 for fibrinogen vs fibrin.
Materials and methods
Reagents
All reagents were obtained from Sigma-Aldrich unless stated otherwise. We previously described our monoclonal antibodies (mAbs) 10E5 (anti-αIIb cap domain),37 7E3 (anti-β3 near the MIDAS),38-40 6D1 (anti-GPIb),41 and 6F1 (anti-α2β1).42 Details on mAb PT25-2, which activates αIIbβ3,43,44 the Thr receptor activating peptide SFLLRN (T6), collagen, collagen-related peptide (CRP), human Thr, ristocetin, soluble human fibrin, fibrinogen, fibrin(ogen) fragments D98 and D-dimer,6 peptides GPRP, RGDW, and KQAGDV, acid-citrate-dextrose, and buffers, are in the supplemental data.
Preparation of human platelets
Healthy volunteers.
This study was approved by The Rockefeller University Institutional Review Board, and it is Helsinki-compliant. Blood was obtained from healthy volunteers who provided informed consent and who had not taken antiplatelet medications during the 2 weeks before blood collection. The blood was anticoagulated with one-sixth volume of acid-citrate-dextrose. Details on preparation of platelet-rich plasma and washed platelets are provided in the supplemental data.
For platelet aggregation studies and studies of the interaction of platelets with polymerizing fibrin, washed platelets were stirred in an aggregometer cuvette and activated with 25 µM T6 or 0.2 U/mL Thr at the indicated time of agonist injection (TI). The time to the onset of the increase in light transmission is reported as TA. αIIbβ3 antagonists (RGDW, γ12-like peptide [KQAGDV], eptifibatide, abciximab, mAb 10E5, and mAb 7E3) were added as indicated 20 minutes prior to initiating aggregation.
Glanzmann thrombasthenia patient.
See the supplemental data for case history. Light transmission aggregometry demonstrated no aggregation in response to ADP, epinephrine, collagen, or arachidonic acid, but a normal response to ristocetin. Flow cytometry demonstrated virtually no binding of mAb 7E3. Reverse transcription polymerase chain reaction of platelet RNA identified a homozygous β3 C575R mutation.
Clot retraction and SEM
Washed platelets (300 μL; 3 × 108/mL) were added to a cuvette containing a mixture of 2 mM CaCl2 and 0.2 U/mL Thr, and clot retraction was monitored by taking photographs of the clots at timed intervals and then analyzing the images for the area of the clot with NIH ImageJ software. For scanning electron microscopy (SEM), clot retraction was performed on a coverslip as described in the supplemental data. The platelet/fibrin suspensions were fixed with 2.5%v glutaraldehyde, 4%v paraformaldehyde, 0.2 M sucrose, and 0.1 M sodium cacodylate, pH 7.4 for 30 minutes at room temperature. They were then deposited on a high-precision glass 1.5 H coverslip, and the samples were dehydrated, dried, sputter-coated with ≈20 nm gold/palladium particles, and imaged with a field-emission scanning electron microscope at 4 kV as described in the supplemental data.
SPR
The interaction of full-length, purified αIIbβ3 with either human fibrinogen or soluble fibrin was assessed by analyzing changes in response units using an SPR instrument (Biacore 8K) as described in the supplemental data. Specific binding between the proteins was calculated by subtracting the reference sensorgram from the sample. Data were analyzed with the Biacore Insight Evaluation 3.0.12.15655 software.
Statistical analysis
Results are expressed as means ± standard deviation (SD) with the number of observations indicated as n. Data were analyzed using GraphPad Prism 6.07. Comparison between samples was determined using the Student t test. Differences were considered significant at P ≤ .05.
Results
The peptide RGDW eliminates the Thr-induced, fibrinogen-mediated, initial wave of platelet aggregation and reveals a delayed wave of increased light transmission (DW) that is mediated by polymerizing fibrin
Because adding Thr to washed platelets results in platelet activation, rapid binding of fibrinogen to αIIbβ3, platelet aggregation (manifested as an initial wave of increased light transmission), initiation of fibrin polymerization, and activation of factor XIII, we sought to inhibit the rapid interactions between platelets and fibrinogen so that we could observe possible later-occurring interactions between platelets and polymerizing fibrin. In dose-response studies, we found that the peptide RGDW profoundly inhibited platelet aggregation of washed platelets initiated by T6 at 5 to 10 µM and essentially eliminated it at concentrations of 50 µM or higher (Figure 1A,C; supplemental Figure 1A). When Thr (0.2 U/mL) was used to activate the platelets, the rapid platelet aggregation response could be eliminated by RGDW at concentrations >5 to 10 µM, and a DW of increased light transmission appeared after several minutes (Figure 1B,C; supplemental Figure 1B). Higher doses of RGDW decreased the slope and the maximal increase in light transmission of the DW, but even at 150 to 200 µM, RGDW failed to inhibit the DW completely (supplemental Figure 1B). Inhibiting fibrin polymerization, as shown by a reduction in γ-γ dimers (supplemental Figure 2A), with the peptide GPRP (5 µM) had no effect on T6-induced platelet aggregation (Figure 1A) and only a minor effect on the initial wave of Thr-induced aggregation in the absence of RGDW, but it eliminated the DW when Thr was combined with RGDW at 150 µM (Figure 1B-C), indicating that the DW requires fibrin polymerization. Immunoblot analysis of samples removed at different times after adding Thr showed that fibrin cross-linking, as judged by the appearance of γ-γ dimers, began by 1 minute, was >50% complete by 5 minutes, and nearly was complete by 10 minutes (Figure 1D).
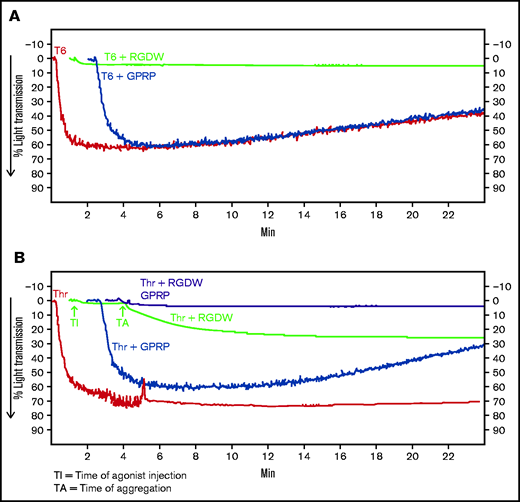
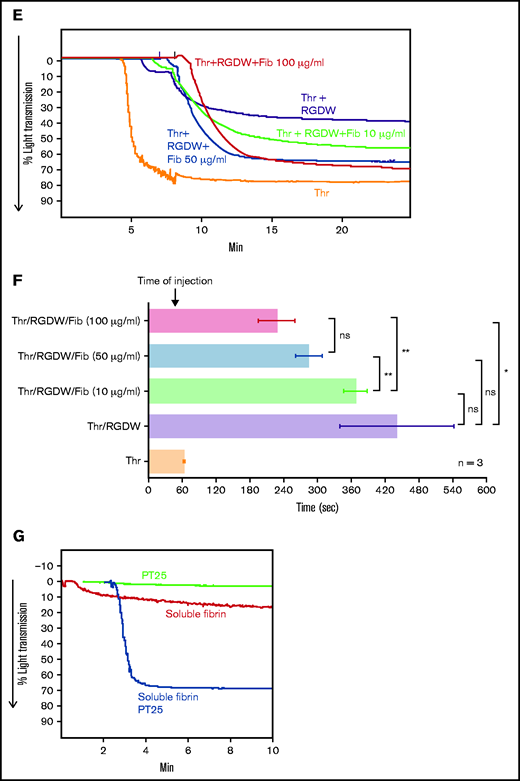
Human platelets treated with the peptide RGDW show a fibrin-mediated delayed wave (DW) of increased light transmission when activated with Thr; fibrinogen enhances the DW, and platelet activation is required for interaction with polymerized fibrin. Washed platelets (2 × 108/mL) were treated with 150 μM RGDW and/or the fibrin polymerization inhibitor Gly-Pro-Arg-Pro (GPRP) for 20 minutes at room temperature and then activated with either (A) 25 μM of a Thr PAR-1 receptor activating peptide (SFLLRN; T6) or (B) 0.2 U/mL Thr in an aggregometer cuvette. Changes in light transmission were measured at 37°C with stirring. (C) Quantitation and statistical analysis of the corresponding maximal change in light transmission (ΔLT) of the T6- or Thr-induced initial wave (<2 minutes after agonist injection) in the presence or absence of RGDW (left panel), and Thr-induced DW (>2 minutes after agonist injection), presented as mean ± SD. Where indicated, samples were treated with human fibrinogen (10, 50, and 100 µg/mL; red dots), GPRP (5 µM; green dots), or mAbs 6D1 or 6F1 (both at 10 µg/mL; blue dots). Data are presented as mean ± SD. Statistical analyses were performed using Student t test between samples tested in the same experiments with and without the indicated intervention (color-coded). *P < .05; ****P < .0001. ns, nonsignificant. (D) In parallel studies, the reaction was stopped at the indicated times by adding one-third volume of 3× sodium dodecyl sulfate (SDS) sample buffer. Samples were subjected to SDS-polyacrylamide gel electrophoresis under reducing conditions and immunoblotted with an mAb specific for the fibrinogen γ-chain. (E) Addition of fibrinogen at 10 µg/mL (blue tracing), 50 µg/mL (purple tracing), 100 µg/mL (black tracing) enhanced the DW. (F) Effect of added fibrinogen on the time to DW onset. Fibrinogen at the indicated concentrations was added to washed platelet samples treated with 150 μM RGDW in an aggregometer cuvette at 37°C with stirring. After 60 seconds, 0.2 U/mL Thr was added (time of injection), and the time of DW onset was recorded. Data were presented as mean ± SD. Significant differences (P < .05) between groups were estimated using Student t test. *P < .05, **P < .01. (G) Washed platelets were treated with 15 μg/mL PT25-2 for 20 minutes at room temperature. Then, soluble fibrin (100 μg/mL) was added, and the change in light transmission was monitored. All tracings and the immunoblot are representative of at least 3 independent experiments.
Human platelets treated with the peptide RGDW show a fibrin-mediated delayed wave (DW) of increased light transmission when activated with Thr; fibrinogen enhances the DW, and platelet activation is required for interaction with polymerized fibrin. Washed platelets (2 × 108/mL) were treated with 150 μM RGDW and/or the fibrin polymerization inhibitor Gly-Pro-Arg-Pro (GPRP) for 20 minutes at room temperature and then activated with either (A) 25 μM of a Thr PAR-1 receptor activating peptide (SFLLRN; T6) or (B) 0.2 U/mL Thr in an aggregometer cuvette. Changes in light transmission were measured at 37°C with stirring. (C) Quantitation and statistical analysis of the corresponding maximal change in light transmission (ΔLT) of the T6- or Thr-induced initial wave (<2 minutes after agonist injection) in the presence or absence of RGDW (left panel), and Thr-induced DW (>2 minutes after agonist injection), presented as mean ± SD. Where indicated, samples were treated with human fibrinogen (10, 50, and 100 µg/mL; red dots), GPRP (5 µM; green dots), or mAbs 6D1 or 6F1 (both at 10 µg/mL; blue dots). Data are presented as mean ± SD. Statistical analyses were performed using Student t test between samples tested in the same experiments with and without the indicated intervention (color-coded). *P < .05; ****P < .0001. ns, nonsignificant. (D) In parallel studies, the reaction was stopped at the indicated times by adding one-third volume of 3× sodium dodecyl sulfate (SDS) sample buffer. Samples were subjected to SDS-polyacrylamide gel electrophoresis under reducing conditions and immunoblotted with an mAb specific for the fibrinogen γ-chain. (E) Addition of fibrinogen at 10 µg/mL (blue tracing), 50 µg/mL (purple tracing), 100 µg/mL (black tracing) enhanced the DW. (F) Effect of added fibrinogen on the time to DW onset. Fibrinogen at the indicated concentrations was added to washed platelet samples treated with 150 μM RGDW in an aggregometer cuvette at 37°C with stirring. After 60 seconds, 0.2 U/mL Thr was added (time of injection), and the time of DW onset was recorded. Data were presented as mean ± SD. Significant differences (P < .05) between groups were estimated using Student t test. *P < .05, **P < .01. (G) Washed platelets were treated with 15 μg/mL PT25-2 for 20 minutes at room temperature. Then, soluble fibrin (100 μg/mL) was added, and the change in light transmission was monitored. All tracings and the immunoblot are representative of at least 3 independent experiments.
The above experiments relied on fibrinogen released from platelets, and so fibrinogen levels were much lower than in plasma (≈15 ug/mL45 compared with 2-4 mg/mL). Adding 10 µg/mL, 50 µg/mL, or 100 µg/mL of fibrinogen to platelets enhanced the DW in a dose-dependent manner (Figure 1C,E). Figure 1F shows that the time to the DW varied inversely as a function of the concentration of fibrinogen, with the greater variability in the absence of added fibrinogen probably reflecting interindividual differences in platelet fibrinogen levels and release of fibrinogen.
Integrin αIIbβ3 activation is required for platelet-fibrin interactions
To assess if integrin activation is required for its interaction with polymerizing fibrin, we used soluble fibrin derived from plasma clots by dissociation using nondenaturing conditions. Under acidic conditions, the fibrin is soluble, but it reassembles into fibrin strands when the solution is neutralized. Immunoblot analysis of the soluble fibrin identified all 3 fibrinogen chains, with >50% of the γ-chains migrating as γ-γ dimers, indicating that it simulates an intermediate time point during fibrin formation and cross-linking (supplemental Figure 2B). Adding soluble fibrin to platelets led to a small (≈15%) increase in light transmission over 10 minutes (Figure 1G). Activating αIIbβ3 with 15 μg/mL PT25-2 dramatically enhanced the increase in light transmission, increasing from ≈15% to 70%.
T6 and Thr primarily induce platelet-platelet interactions, whereas Thr/RGDW primarily induces platelet-fibrin interactions.
SEM of washed platelets at 30 seconds and 2 minutes after treatment with 25 µM T6 showed primarily platelet-platelet interactions forming variably sized aggregates, whereas platelets treated with 0.2 U/mL Thr for the same period of time showed a mixture of platelet aggregation and platelet interaction with polymerizing fibrin (Figure 2A). In the presence of RGDW, however, there were few platelet-platelet interactions and a predominance of platelet-fibrin interactions. In the Thr/RGDW group, at high magnification (Figure 2A right 2 panels; Figure 2B), one could see that the fibrin strands had extended lengths of interactions with the surface of single platelets, with some strands interacting >1 µm or more. In addition, Thr/RGDW-treated platelets exhibited a rounder shape and fewer filopodia and lamellipodia than their Thr-treated counterparts. At 4 minutes after Thr treatment in the absence of RGDW (Figure 2B), there was extensive, tight, platelet-platelet aggregate formation and extensive platelet-fibrin interactions. In contrast, at 4 minutes after the onset of the DW with Thr/RGDW treatment, there was a predominance of single platelet-fibrin interactions.
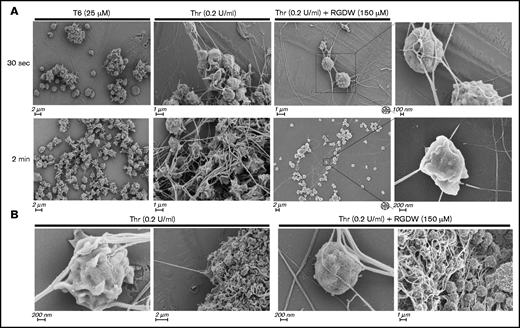
SEM demonstrates that RGDW inhibits platelet-platelet interactions, but not platelet interactions with polymerizing fibrin in the DW. Representative SEM images of washed platelets (2 × 108/mL) stimulated in the aggregometer with the indicated agonist, with or without pretreatment with 150 μM RGDW. (A) Samples fixed 30 seconds (top row) or 2 minutes (lower row) after initiation of aggregation or onset of the DW. (B) Samples fixed 4 minutes after activation with Thr alone (left 2 panels) or Thr with RGDW (right 2 panels).
SEM demonstrates that RGDW inhibits platelet-platelet interactions, but not platelet interactions with polymerizing fibrin in the DW. Representative SEM images of washed platelets (2 × 108/mL) stimulated in the aggregometer with the indicated agonist, with or without pretreatment with 150 μM RGDW. (A) Samples fixed 30 seconds (top row) or 2 minutes (lower row) after initiation of aggregation or onset of the DW. (B) Samples fixed 4 minutes after activation with Thr alone (left 2 panels) or Thr with RGDW (right 2 panels).
Platelet glycoprotein receptors GPVI, α2β1, and GPIb play relatively small, if any, roles in mediating the DW
To assess the role of GPVI on the DW, we incubated washed platelets with 2 different inhibitors of platelet aggregation initiated by the interaction of CRP with GPVI, PP2 (100 mM), and Syk inhibitor IV (1 mM). Both inhibitors essentially eliminated CRP-induced aggregation of washed platelets but had minimal effect on the DW (Figure 3A). Consistent with a lack of GPVI ligand engagement, platelet samples obtained during the DW did not demonstrate evidence of signaling downstream of GPVI ligand engagement as judged by Syk or PLCγ2 phosphorylation (supplemental Figure 3). Antibody 6F1 (10 μg/mL), which inhibits α2β1-mediated adhesion to collagen,42 had little if any impact on the DW (Figures 3B and 1C). Antibody 6D1 (10 μg/mL), which is a potent inhibitor of von Willebrand factor binding to GPIb,41 had a modest effect on the DW, reducing the maximal amplitude of the DW by 5% (Figures 3B and 1C).
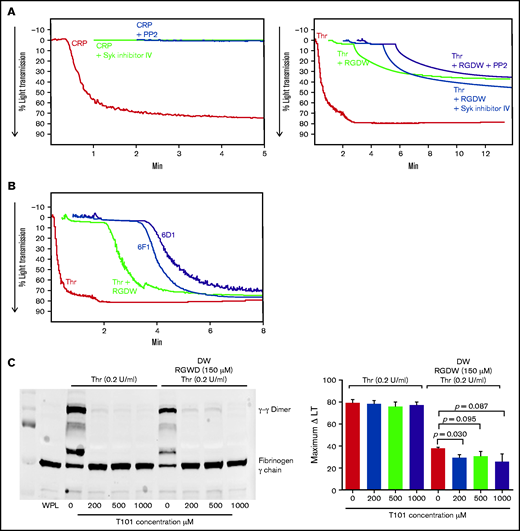
Inhibition of Syk and Src kinases, GPIb, integrin α2β1, or FXIII-mediated fibrin cross-linking, has little or no effect on the time to onset or maximal change in light transmission of the DW. RGDW (150 μM)-treated washed platelets (2 × 108/mL) were incubated for 20 minutes at RT with the following: (A) 100 μM of the Src kinase inhibitor PP2 or 1 μM of the Syk kinase inhibitor VI, or (B) 10 μg/mL of the GPIb-blocking mAb 6D1 or the α2β1-blocking mAb 6F1. Presented tracings are representative of 3 independent experiments. (C) Washed platelets in the absence or presence of RGDW (150 μM) were treated with the FXIII transglutaminase inhibitor T101 at the indicated concentrations 5 minutes prior to adding Thr (0.2 U/mL). Left panel: immunoblot data using a mAb specific for fibrinogen γ-chain of samples solubilized with SDS sample buffer under reducing conditions after 15 minutes of activation. Right panel: corresponding maximal ΔLT of the DW in 3 independent experiments, presented as mean ± SD. WPL, washed platelet lysate.
Inhibition of Syk and Src kinases, GPIb, integrin α2β1, or FXIII-mediated fibrin cross-linking, has little or no effect on the time to onset or maximal change in light transmission of the DW. RGDW (150 μM)-treated washed platelets (2 × 108/mL) were incubated for 20 minutes at RT with the following: (A) 100 μM of the Src kinase inhibitor PP2 or 1 μM of the Syk kinase inhibitor VI, or (B) 10 μg/mL of the GPIb-blocking mAb 6D1 or the α2β1-blocking mAb 6F1. Presented tracings are representative of 3 independent experiments. (C) Washed platelets in the absence or presence of RGDW (150 μM) were treated with the FXIII transglutaminase inhibitor T101 at the indicated concentrations 5 minutes prior to adding Thr (0.2 U/mL). Left panel: immunoblot data using a mAb specific for fibrinogen γ-chain of samples solubilized with SDS sample buffer under reducing conditions after 15 minutes of activation. Right panel: corresponding maximal ΔLT of the DW in 3 independent experiments, presented as mean ± SD. WPL, washed platelet lysate.
Inhibiting factor XIII-mediated fibrin crosslinking has little impact on the DW
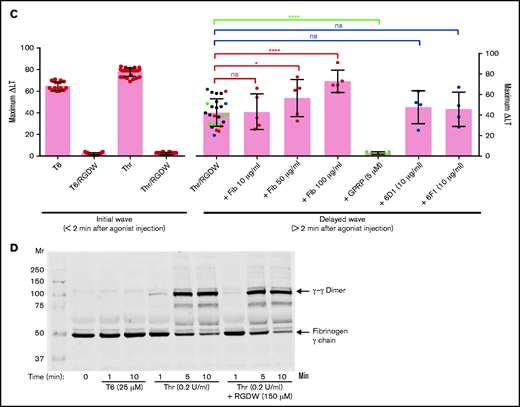
We used the factor XIII inhibitor T10146 to test whether cross-linking is required for the platelet-fibrin interactions leading to the DW. Increasing doses of T101 reduced fibrin cross-linking by >95% as judged by the formation of γ-γ dimers (Figure 3C; supplemental Figure 4A). At 200, 500, and 1000 µM, it decreased the DW by 24% (P = .03), 19% (P = .095), and 32% (P = .087; n = 3) (Figure 3C; supplemental Figure 4B).
Integrin αIIbβ3 is the dominant receptor mediating the DW
To assess the role of αIIbβ3 in the DW, we studied a patient with Glanzmann thrombasthenia whose platelets have virtually no surface expression of the receptor. As shown in Figure 4A, unlike the control platelets, which aggregated in response to both T6 and Thr and had a prominent DW when stimulated with Thr and RGDW, the patient’s platelets did not respond to any of these stimuli and did not demonstrate a DW. Even in the presence of preformed soluble fibrin, platelet activation by either T6 (Figure 4B) or PT25-2 (Figure 4C) failed to produce an increase in light transmission. Concordantly, SEM images demonstrated that platelets from the Glanzmann thrombasthenia patient did not interact with fibrin (Figure 4D).
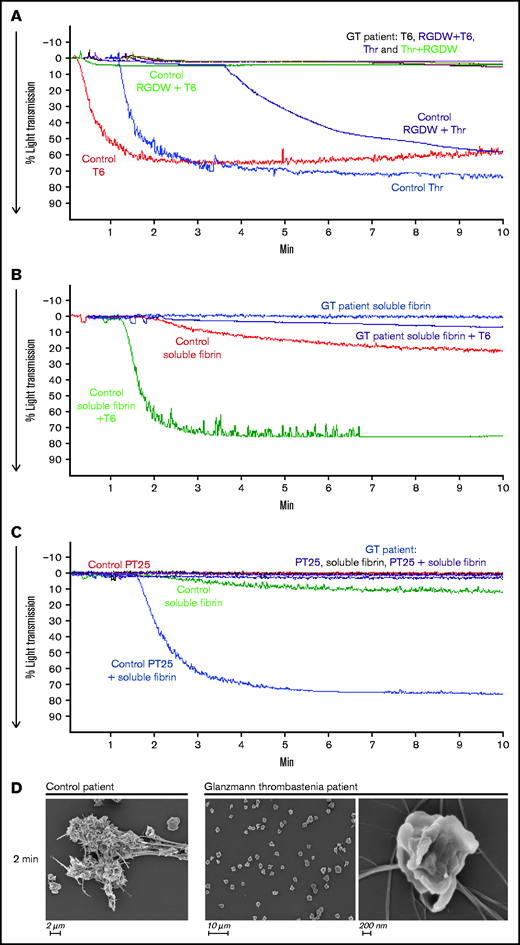
Platelets from a patient with Glanzmann thrombasthenia do not aggregate with T6 or Thr, do not demonstrate a DW in response to Thr + RGDW, and do not interact with soluble fibrin when activated with T6 or mAb PT25-2. Fibrinogen (50 µg/mL) was added to washed platelets (2 × 108/mL) from a healthy donor (Control) or a patient with Glanzmann thrombasthenia (GT), and then the platelets were treated with the following: (A) T6 (25 μM), Thr (0.2 U/mL), T6 (25 μM) + RGDW (150 μM), or Thr (0.2 U/mL) + RGDW (150 μM). (B) Soluble fibrin (100 μg/mL) was added to washed control or patient platelets and then the platelets were either untreated or treated with 25 μM T6. (C) Washed control or patient platelets (2 × 108/mL) were treated with 15 μg/mL PT25-2 for 20 minutes at room temperature, and then soluble fibrin (100 μg/mL) was added and the change in light transmission was monitored. (D) SEM of samples from a healthy donor and the GT patient 2 minutes after the onset of the DW in the healthy donor.
Platelets from a patient with Glanzmann thrombasthenia do not aggregate with T6 or Thr, do not demonstrate a DW in response to Thr + RGDW, and do not interact with soluble fibrin when activated with T6 or mAb PT25-2. Fibrinogen (50 µg/mL) was added to washed platelets (2 × 108/mL) from a healthy donor (Control) or a patient with Glanzmann thrombasthenia (GT), and then the platelets were treated with the following: (A) T6 (25 μM), Thr (0.2 U/mL), T6 (25 μM) + RGDW (150 μM), or Thr (0.2 U/mL) + RGDW (150 μM). (B) Soluble fibrin (100 μg/mL) was added to washed control or patient platelets and then the platelets were either untreated or treated with 25 μM T6. (C) Washed control or patient platelets (2 × 108/mL) were treated with 15 μg/mL PT25-2 for 20 minutes at room temperature, and then soluble fibrin (100 μg/mL) was added and the change in light transmission was monitored. (D) SEM of samples from a healthy donor and the GT patient 2 minutes after the onset of the DW in the healthy donor.
Inhibition of the DW correlates with inhibition of clot retraction
To assess whether the interaction between platelets and polymerizing fibrin in our assay is similar to the interaction between platelets and fibrin that results in clot retraction, we compared the results of our DW assay studies with those found with clot retraction. We found a correspondence between the impact of RGDW in delaying, but not preventing clot retraction (Figure 5A). SEM images demonstrated that in the absence of RGDW, as with Thr-induced platelet aggregation, there are masses of aggregated platelets interacting with thick fibrin strands connecting the platelet aggregates. In the presence of RGDW, there were smaller platelet aggregates, more single platelet-fibrin interactions, and a denser web of fibrin strands (Figure 5B).

As with the DW, RGDW only partially inhibits clot retraction; αIIbβ3 activation is required for retraction of preformed fibrin. (A) Washed platelets (3 × 108/mL) were treated with the RGDW (150 µM) for 20 minutes at room temperature and then transferred to an aggregometer cuvette containing 0.2 U/mL Thr and 2 mM CaCl2 to initiate clot retraction. The area of the clot is shown underneath each photograph and expressed in square pixels. (B) SEM images of clot retraction after 15 minutes. (C) Washed platelets were untreated (sample 1) or stimulated to undergo clot retraction with the following: Thr + CaCl2 (sample 2), or soluble fibrin + CaCl2 alone (sample 3), or in combination with 15 μg/mL of the mAb PT25-2 (sample 4), or 25 μM T6 (sample 5). Photographs of the time course of clot retraction are shown. The area of the clot is shown underneath each photograph and expressed in square pixels. Data shown in panels A and C are representative of at least 3 independent experiments.
As with the DW, RGDW only partially inhibits clot retraction; αIIbβ3 activation is required for retraction of preformed fibrin. (A) Washed platelets (3 × 108/mL) were treated with the RGDW (150 µM) for 20 minutes at room temperature and then transferred to an aggregometer cuvette containing 0.2 U/mL Thr and 2 mM CaCl2 to initiate clot retraction. The area of the clot is shown underneath each photograph and expressed in square pixels. (B) SEM images of clot retraction after 15 minutes. (C) Washed platelets were untreated (sample 1) or stimulated to undergo clot retraction with the following: Thr + CaCl2 (sample 2), or soluble fibrin + CaCl2 alone (sample 3), or in combination with 15 μg/mL of the mAb PT25-2 (sample 4), or 25 μM T6 (sample 5). Photographs of the time course of clot retraction are shown. The area of the clot is shown underneath each photograph and expressed in square pixels. Data shown in panels A and C are representative of at least 3 independent experiments.
To assess whether clot retraction, like the DW, requires integrin activation, we added soluble fibrin to washed platelets in the absence and presence of T6 or PT25-2. In the absence of either activator, there was no clot retraction. In contrast, adding either T6 or the αIIbβ3-selective and specific activator PT25-2 resulted in clot retraction (Figure 5C).
Small-molecule and mAb αIIbβ3 antagonists variably inhibit the DW, whereas D-dimer and D98 both inhibit the DW
The γ-chain 406-411 KQAGDV peptide, which mediates the interaction of fibrinogen with αIIbβ3 by attaching to the αIIbβ3 RGD-binding pocket,25 dramatically inhibited T6-induced platelet aggregation at 100 to 200 µM, and partially inhibited T6-induced binding of the mAb PAC-1 (which has higher affinity for activated αIIbβ3 than does fibrinogen, and which binds to the RGD-binding pocket),47 but had no effect on the DW at concentrations up to 500 µM (Figure 6A-C). The RGDW peptide was similarly highly potent in inhibiting T6-induced platelet aggregation, less potent at inhibiting PAC-1 binding, and least potent in inhibiting the DW. Eptifibatide, a high-affinity αIIbβ3 antagonist that also binds in the RGD-binding pocket,48 nearly eliminated T6-induced platelet aggregation at just 5 µM, fully eliminated PAC-1 binding at 100 µM, but required 500 µM for near-complete inhibition of the DW (Figure 6A-C).
![Integrin αIIbβ3 antagonists differentially affect the DW and platelet-polymerizing fibrin interactions, and D-dimer and D98 inhibit the DW. Washed platelets (2 × 108/mL) were treated for 20 minutes at room temperature with increasing doses of the peptides RGDW or KQAGDV, eptifibatide, the mAbs 7E3 or 10E5, or the chimeric mAb 7E3 Fab fragment abciximab. After incubation, platelets were treated with 25 μM T6 (A,D) or 0.2 U/mL Thr (C,E) and stirred in an aggregometer at 37°C. The change in light transmission 15 minutes after adding the agonist was recorded as the maximal ΔLT. (B) In parallel studies, untreated and αIIbβ3 antagonist-treated platelets (2 × 106/mL) were activated with T6 (25 μM) under static conditions without stirring for 5 minutes and incubated with Alexa488-labeled PAC-1 for 20 minutes, after which platelet-associated fluorescence was analyzed by flow cytometry. The data are reported as the mean ± SD inhibition of the geometric mean fluorescence in the absence of each antagonist. (F) SEM of samples of washed platelets 2 minutes after the onset of the DW in the presence of either mAb 7E3 or 10E5. (G) Adding purified D-dimer or D98 at 200 µg/mL to the washed platelets inhibited the DW. One of 3 similar experiments with D-dimer and D98 (mean DW ± SD without and with D-dimer equal 42 ± 6.5 and 5.3 ± 3.2 [P < .005]; mean DW ± SD without and with D98 equal 42 ± 6.5 and 1.7 ± 0.5 [P < .005]).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/20/10.1182_bloodadvances.2021005142/2/m_advancesadv2021005142f6a.png?Expires=1769142706&Signature=hbv3OJP0P3EH3OvpSO8DW46vxWmNOl1lWLzZ2-Xn-lu6aZF8m2bQkewBXeXln7rJPIa9nDiR4kyAVhpBT5z-q9TcH1YrhYGBgFUNyIY-SxURsWU4D~FreQAy0DAg7NbbW8ZabiN088XVPu1hpXizukDQ3d3ytZhlFtgF9TOAAmrFWB-e6UnFk4EDPrB-fF3yThSL39xEorNDek8kK-rETDQVtqj-CadYaO7NhKqnbsWtvJCh7ENQWKZVpIulXXjEw1eM6AMXIFs-9atx8vVvHzicAaVAy77OrJOn-ERJc-EjZH~DTj19oZ-4LuNrSTpKVHe-AJTAJdz~nf66-kpC4g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![Integrin αIIbβ3 antagonists differentially affect the DW and platelet-polymerizing fibrin interactions, and D-dimer and D98 inhibit the DW. Washed platelets (2 × 108/mL) were treated for 20 minutes at room temperature with increasing doses of the peptides RGDW or KQAGDV, eptifibatide, the mAbs 7E3 or 10E5, or the chimeric mAb 7E3 Fab fragment abciximab. After incubation, platelets were treated with 25 μM T6 (A,D) or 0.2 U/mL Thr (C,E) and stirred in an aggregometer at 37°C. The change in light transmission 15 minutes after adding the agonist was recorded as the maximal ΔLT. (B) In parallel studies, untreated and αIIbβ3 antagonist-treated platelets (2 × 106/mL) were activated with T6 (25 μM) under static conditions without stirring for 5 minutes and incubated with Alexa488-labeled PAC-1 for 20 minutes, after which platelet-associated fluorescence was analyzed by flow cytometry. The data are reported as the mean ± SD inhibition of the geometric mean fluorescence in the absence of each antagonist. (F) SEM of samples of washed platelets 2 minutes after the onset of the DW in the presence of either mAb 7E3 or 10E5. (G) Adding purified D-dimer or D98 at 200 µg/mL to the washed platelets inhibited the DW. One of 3 similar experiments with D-dimer and D98 (mean DW ± SD without and with D-dimer equal 42 ± 6.5 and 5.3 ± 3.2 [P < .005]; mean DW ± SD without and with D98 equal 42 ± 6.5 and 1.7 ± 0.5 [P < .005]).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/20/10.1182_bloodadvances.2021005142/2/m_advancesadv2021005142f6b.png?Expires=1769142706&Signature=IWUFzhKrBADLRUKVuBc3NEtPpfJSj1noknIl3~qokofuu2lTyARNlZfGnb7Ga44MqPmxGxlNeifxFQrqpugMmJosfhGYqiJYwpS2xw~Zkf2GJ00~bXVBLke1UO4WpKBOCZgNmhIZNAMNwcRASPj6YgrqwCEn2oUwW5hdeL4tJ50vA3pECliMNYLbwAbmxoC6MsTxVgrrkpOFEJkHkVYobLdXCSIbAtr~osmg~i1rIiQ3mCwBgTqgAmGh~upsdwkpfY9uH4vy9IVcJ9b~pmM8E2PGJTdkJoCL0dY4PDYjzWwO0zzr6EmG2tFrm2XQ8Rm8H-jYWbJrYEytKtaPQWL~IA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Integrin αIIbβ3 antagonists differentially affect the DW and platelet-polymerizing fibrin interactions, and D-dimer and D98 inhibit the DW. Washed platelets (2 × 108/mL) were treated for 20 minutes at room temperature with increasing doses of the peptides RGDW or KQAGDV, eptifibatide, the mAbs 7E3 or 10E5, or the chimeric mAb 7E3 Fab fragment abciximab. After incubation, platelets were treated with 25 μM T6 (A,D) or 0.2 U/mL Thr (C,E) and stirred in an aggregometer at 37°C. The change in light transmission 15 minutes after adding the agonist was recorded as the maximal ΔLT. (B) In parallel studies, untreated and αIIbβ3 antagonist-treated platelets (2 × 106/mL) were activated with T6 (25 μM) under static conditions without stirring for 5 minutes and incubated with Alexa488-labeled PAC-1 for 20 minutes, after which platelet-associated fluorescence was analyzed by flow cytometry. The data are reported as the mean ± SD inhibition of the geometric mean fluorescence in the absence of each antagonist. (F) SEM of samples of washed platelets 2 minutes after the onset of the DW in the presence of either mAb 7E3 or 10E5. (G) Adding purified D-dimer or D98 at 200 µg/mL to the washed platelets inhibited the DW. One of 3 similar experiments with D-dimer and D98 (mean DW ± SD without and with D-dimer equal 42 ± 6.5 and 5.3 ± 3.2 [P < .005]; mean DW ± SD without and with D98 equal 42 ± 6.5 and 1.7 ± 0.5 [P < .005]).
Integrin αIIbβ3 antagonists differentially affect the DW and platelet-polymerizing fibrin interactions, and D-dimer and D98 inhibit the DW. Washed platelets (2 × 108/mL) were treated for 20 minutes at room temperature with increasing doses of the peptides RGDW or KQAGDV, eptifibatide, the mAbs 7E3 or 10E5, or the chimeric mAb 7E3 Fab fragment abciximab. After incubation, platelets were treated with 25 μM T6 (A,D) or 0.2 U/mL Thr (C,E) and stirred in an aggregometer at 37°C. The change in light transmission 15 minutes after adding the agonist was recorded as the maximal ΔLT. (B) In parallel studies, untreated and αIIbβ3 antagonist-treated platelets (2 × 106/mL) were activated with T6 (25 μM) under static conditions without stirring for 5 minutes and incubated with Alexa488-labeled PAC-1 for 20 minutes, after which platelet-associated fluorescence was analyzed by flow cytometry. The data are reported as the mean ± SD inhibition of the geometric mean fluorescence in the absence of each antagonist. (F) SEM of samples of washed platelets 2 minutes after the onset of the DW in the presence of either mAb 7E3 or 10E5. (G) Adding purified D-dimer or D98 at 200 µg/mL to the washed platelets inhibited the DW. One of 3 similar experiments with D-dimer and D98 (mean DW ± SD without and with D-dimer equal 42 ± 6.5 and 5.3 ± 3.2 [P < .005]; mean DW ± SD without and with D98 equal 42 ± 6.5 and 1.7 ± 0.5 [P < .005]).
We then tested 2 different mAbs with nanomolar affinities for αIIbβ3 that are potent inhibitors of fibrinogen binding to activated platelets and platelet aggregation. The mAb 7E3 binds primarily near the RGD-binding pocket in β3,37-40 whereas mAb 10E5 binds primarily to the αIIb cap domain.37,48 Both antibodies nearly completely inhibited platelet aggregation initiated by T6 at 10 µg/mL (Figure 6D; supplemental Figure 1C,E), but they showed dramatically different effects on the DW, with 7E3 producing ≈75% inhibition at 50 to 100 µg/mL, and 10E5 failing to inhibit the DW at concentrations up to 100 µg/mL (Figure 6E; supplemental Figure 1D,F). SEM analysis confirmed that 7E3 at 100 µg/mL dramatically inhibited the interaction of platelet with fibrin, whereas 10E5 at the same concentration did not inhibit the interaction (Figure 6F). Of particular note, although mAb 7E3 inhibited the DW, abciximab, the chimeric Fab fragment of 7E3, which shares the same antibody binding site, and dramatically inhibits T6-induced platelet aggregation at 5 µg/mL (Figure 6D), produced only minimal inhibition of the DW at concentrations up to 100 µg/mL (Figure 6E).
To localize the binding site for activated platelets on polymerizing fibrin, we added purified D-dimer at 200 µg/mL and found that it dramatically inhibited the DW, as did D98 at the same concentration (Figure 6G).
SPR studies with purified, monomeric αIIbβ3 show similar affinities for immobilized fibrinogen and fibrin, in both the absence and presence of priming by mAb PT25-2
In order to assess the relative contributions of affinity and avidity differences in the interaction of αIIbβ3 with fibrinogen and fibrin, we measured the affinity of purified, monomeric αIIbβ3 to immobilized fibrinogen and immobilized soluble fibrin in the absence and presence of the activating mAb PT25-2 (Figure 7A-B). We found that the binding of αIIbβ3 to both fibrinogen and fibrin best fit a heterogeneous ligand binding model (χ2 values between 3 and 27), which is based on the interaction between 1 analyte, in our data αIIbβ3, with 2 different ligands (or potentially a single ligand with 2 different conformations), in our case either fibrinogen or fibrin.49 The data fit a 1:1 model least well (χ2 values between 65 and 619), whereas the fit to a bivalent analyte model was similar to, but not quite as good as, the heterogeneous model (χ2 values between 4 and 601) (supplemental Figure 5A-B). The ka1 for fibrinogen (1.21 × 104) was similar to the ka1 for fibrin (1.34 × 104), as were their kd1 values (2.79 × 10−3 and 1.96 × 10−3, respectively), resulting in very similar Kd1 values (2.31 × 10−7 and 1.47 × 10−7, respectively). Both the ka2 and kd2 values for both ligands were substantially higher than the ka1 and kd1 values, but the values for fibrinogen and fibrin were again similar, resulting in similar KD2 values (5.02 × 10−7 and 3.79 × 10−7). Priming αIIbβ3 with PT25-2 increased the ka1 values for both fibrinogen and fibrin approximately threefold without materially changing the kd1 values, resulting in Kd1 values of 8.50 × 10−8 and 4.78 × 10−8, respectively. Priming lowered both the ka2 and the kd2 values to similar extents, resulting in similar KD2 values (2.31 × 10−7 and 1.37 × 10−7, respectively). In the presence of either mAb 7E3 or mAb 10E5, the binding of αIIbβ3 to fibrinogen (response units) was dramatically decreased (supplemental Figure 6A). In contrast, as with their differential impact on the DW, mAb 7E3 was better able to inhibit the binding of αIIbβ3 to fibrin (supplemental Figure 6B). A repeat experiment gave very similar results.
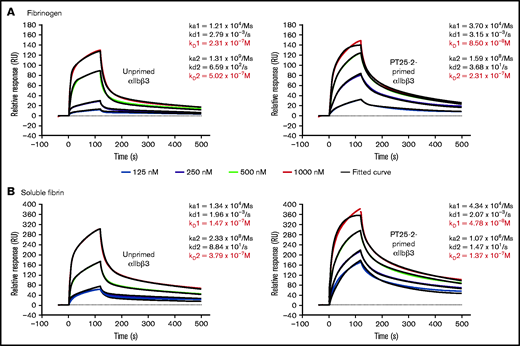
SPR analysis demonstrates that αIIbβ3 has similar affinity for fibrinogen and fibrin. Purified human αIIbβ3 binding to (A) immobilized fibrinogen and (B) soluble fibrin. Where indicated, purified integrin was treated with 15 µg/mL PT25-2 at room temperature for 20 minutes. Kinetic values were obtained by fitting data to a heterogenous ligand interaction model.
SPR analysis demonstrates that αIIbβ3 has similar affinity for fibrinogen and fibrin. Purified human αIIbβ3 binding to (A) immobilized fibrinogen and (B) soluble fibrin. Where indicated, purified integrin was treated with 15 µg/mL PT25-2 at room temperature for 20 minutes. Kinetic values were obtained by fitting data to a heterogenous ligand interaction model.
Discussion
Our data demonstrate that the interaction of Thr-activated platelets with fibrinogen, and the ensuing platelet aggregation, can be differentiated from the interaction of Thr-activated platelets with polymerizing fibrin by including the RGDW peptide; the latter eliminates the initial platelet aggregation response, but leaves intact a DW of platelet interactions with fibrin, which in turn can be eliminated by a peptide (GPRP) that prevents fibrin polymerization.50 We found that the DW has an absolute requirement for αIIbβ3 because it was absent when platelets from a patient with Glanzmann thrombasthenia were substituted for control platelets. The DW could be inhibited by the potent αIIbβ3 antagonists eptifibatide and mAb 7E3, but much higher concentrations were required than those needed to inhibit fibrinogen-mediated platelet aggregation induced by T6. To differentiate whether the requirement for higher concentrations of the inhibitors was due to αIIbβ3 having higher affinity or avidity, or both, for fibrin vs fibrinogen, we performed SPR studies in which monomeric αIIbβ3 in solution bound to either immobilized fibrinogen or soluble fibrin. We found that the binding to each best fit a heterogeneous ligand binding model and that the ka1, kd1, ka2, and kd2 values were similar for both fibrinogen and fibrin, suggesting that the affinity of the receptor was similar for both proteins. By inference, this suggests that the stronger interaction with fibrin reflects increased avidity due to the simultaneous engagement of multiple αIIbβ3 receptors. Our SEM studies of the interaction of platelets with polymerizing fibrin are consistent with this interpretation because platelets make intimate contact with extended regions of the polymerizing fibrin strands, in some cases several microns in length. Whereas αIIbβ3 receptors are only ≈200 Å apart, on average, on the platelet surface, the interacting polymerizing fibrin strands likely contact many αIIbβ3 receptors. This would translate into dramatically enhanced avidity as the cumulative off-rate is the product of the individual off-rates of the interactions.35 Although our SPR data fit the heterogeneous model, in which there are 2 different ligands and a single analyte, they also fit the bivalent analyte model, in which the analyte makes 2 interactions, nearly as well. These results are consistent with the complex pattern of fibrinogen and fibrin interaction with αIIbβ3 in which there are multiple steps and interacting sites on the receptor and ligand.
The difference in the ability of mAb 7E3 and its chimeric Fab fragment, abciximab, to inhibit the DW is striking because they share the same antibody binding site.51 We previously showed that mAb 7E3 binds bivalently to αIIbβ3 on platelets,52 and thus, its off-rate will be the product of the off-rates for each binding site. In contrast, as a Fab fragment, abciximab can only make monovalent contact and thus will have lower avidity even though the affinity of the individual interactions should be the same. Thus, the bivalent binding of mAb 7E3 is sufficient to overcome the greater avidity of the interaction of αIIbβ3 with polymerizing fibrin than with fibrinogen. Of note, Collet et al also found that that abciximab did not prevent the interaction of platelets with fibrin during in vitro Thr-induced clotting, but by inhibiting platelet-platelet interactions, it facilitated lysis by tissue plasminogen activator.7,53,54 Similar results were found by Braaten et al using an RGD peptide.55
Surprisingly, mAb 10E5, a potent inhibitor of fibrinogen binding to αIIbβ3 (KD ≈ 16 nM), and of platelet aggregation initiated by multiple agonists,37 did not inhibit the DW even at high concentrations. This raises the possibility that fibrin binding is less dependent on interacting with the αIIb cap domain, which is 10E5’s epitope,48 than the β3 specificity-determining loop and β1-α1 loop in the I domain, which is where 7E3 binds.39,40 These new data are in accord with our previous finding that 10E5 is also less potent at inhibiting platelet adhesion to D-dimer and clot retraction.6
We previously reviewed the potential for the fibrinogen γ404-411 sequence to mediate the interaction of αIIbβ3 with D-dimer,6 and the same analysis applies to cross-linked fibrin because in both cases the Lys406 will not be available for an ionic interaction with αIIb Asp224. Thus, it is possible that the γ407-411 peptide can bind to the β3 subunit via coordination of the MIDAS Mg2+ by the Asp410 carboxyl group and via an interaction of Val411 (through an intermediary water molecule) with the ADMIDAS (Adjacent to MIDAS) Ca2+,25 if the peptide retains sufficient accessibility and flexibility. The ability of fibrinogen fragment D98, which lacks the γ407-411 sequence, to inhibit both clot retraction6 and the DW does not exclude a potential role for γ407-411, because D98’s inhibition may reflect it containing a necessary ancillary binding site for fibrin binding, or allosteric inhibition if it interacts with the polymerizing fibrin.
Litvinov et al used optical trap-based force spectroscopy to study the interaction of αIIbβ3 (activated with MnCl2) with fibrinogen or fibrin monomer.31 They found that the interaction with fibrin had higher binding probability and higher rupture force but noted that a substantial percentage of the αIIbβ3-fibrin interactions was probably multimolecular. Thus, they could not formally differentiate between affinity and avidity effects. They also found that an RGD peptide, eptifibatide, and abciximab were less effective in inhibiting the interaction with fibrin compared with fibrin, which is in accord with our finding with the DW.
We found little evidence of a major contribution of either GPVI or GPIb to the DW because neither inhibitor of GPVI signaling nor GPIb binding to von Willebrand factor had a strong impact. Moreover, we could not identify evidence of GPVI signaling as a result of the DW. Our data differ from those of others who used different assays,14-16 and we did not perform studies of the role of GPIb under controlled high shear force conditions, so additional studies are required to assess the relative contributions of these other receptors.
We found that platelets showed relatively little interaction with soluble fibrin unless activated with T6 or PT25-2. These data are in accord with our previous data demonstrating that platelet activation is required for adhesion to D-dimer,6 and data from multiple groups that platelet activation is required for clot retraction.56-58 We previously found that D-dimer can inhibit clot retraction as can a polyclonal antibody to D-dimer.6 In the present study, we also found that D-dimer can inhibit the DW, another similarity between these phenomena.
There are conflicting data on whether FXIII activity is required for platelet-mediated clot retraction, which requires platelet interactions with polymerizing fibrin and force generation.59-66 Results with mouse blood also vary, with Kasahara et al reporting that clot retraction requires cross-linked fibrin,67,68 whereas Kattula et al found equivalent retraction in wild-type and F13a−/− mice.46 In the presence of red cells, the effect of FXIII on entrapping red cells via α-polymer formation complicates the analysis, but this does not bear on the effect of FXIII on the interaction of αIIbβ3 with fibrin.46,69,70 Although we found evidence of fibrin cross-linking before the DW, inhibiting FXIII-mediated cross-linking by >95% reduced the DW by only 32%.
Our data have potentially important clinical significance. In particular, the higher avidity of fibrin for αIIbβ3 than fibrinogen means that the current αIIbβ3 antagonists may not be effective in inhibiting platelet fibrin interactions, even when they dramatically inhibit platelet-fibrinogen interactions and platelet aggregation. For example, eptifibatide reaches plasma levels of at most 2 to 3 µM,71 which is enough to profoundly inhibit T6-induced platelet aggregation and PAC-1 binding, but only inhibit the DW by ∼50%. Abciximab is even less potent in inhibiting the DW at the peak concentration it reaches in vivo after bolus administration.72 It is thus possible that new αIIbβ3 antagonists with very high affinity may be able inhibit platelet-fibrin interactions, and potentially be more effective as antithrombotic agents.
Acknowledgments
The authors thank Carolina Adura and Lavoisier Ramos-Espiritu of the High-Throughput and Spectroscopy Resource Center of The Rockefeller University for their support and technical advice for SPR studies, Hilda Amalia Pasolli and Nadine Soplop from the Electron-Microscopy Resource Center of The Rockefeller University for their SEM services and for their valuable technical support, and The Rockefeller University Proteomics Resource Center for peptide synthesis. The authors give special thanks to Jihong Li for her advice and support throughout this study and to Suzanne Rivera for outstanding secretarial and administrative support.
This work was supported in part by National Institutes of Health (NIH), National Heart, Lung, and Blood Institute grant 19278, National Center for Advancing Translational Sciences (NCATS) grant UL1 TR001866, National Institutes of Health (NIH) Clinical and Translational Science Award (CTSA) program, and a pilot project grant supported by the Shapiro-Silverberg Fund at Rockefeller University.
Authorship
Contribution: L.B. designed, performed, and analyzed functional experiments and wrote the manuscript; S.L. performed and analyzed SEM experiments; O.B. contributed the clinical assessment of Glanzmann thrombasthenia patient and wrote the manuscript; J.P. analyzed SPR data and wrote the manuscript; B.C. designed, oversaw, and analyzed the data and had primary responsibility for writing the manuscript.
Conflict-of-interest disclosure: B.C. receives royalty payments from the Research Foundation of the State University of New York based on the sales of abciximab. The remaining authors declare no competing financial interests.
Correspondence: Barry Coller, Allen and Frances Adler Laboratory of Blood and Vascular Biology, Hospital Building, Room 617, The Rockefeller University, 1230 York Ave, New York, NY 10065; e-mail: collerb@rockefeller.edu.

