

Key Points
The recipients’ preformed alloantibodies may initiate TRALI reaction in recipients transfused with soluble antigen.
The antibody interaction with absorbed antigen on ECs leads to endothelial barrier dysfunction and lung injury.

Abstract
Transfusion-related lung injury (TRALI) is a serious side effect of blood transfusion. Exclusion of antibody carriers from the donor pool has significantly decreased the number of cases, but TRALI remains the leading cause of transfusion-related morbidity and mortality in industrialized countries. Here, we show that proteins released from donor cells during processing of blood components are capable of inducing a new type of reverse TRALI when transfused to preimmunized recipients. First, we show that soluble neutrophil surface protein CD177 in complex with proteinase 3 (sCD177/PR3) is not only present in human plasma but also in packed red blood cell (PRBC) supernatant. Filtration or storage enhances the concentration of sCD177/PR3 in PRBCs. Second, we show that sCD177/PR3 specifically binds to PECAM-1 on stimulated (but not on unstimulated) endothelial cells (ECs). Third, we provide evidence that the sCD177/PR3/PECAM-1 complex is functional. In the presence of monoclonal or human antibodies against CD177 or PR3, ECs produce reactive oxygen species and become apoptotic. Albumin flux through an EC monolayer increases significantly whenever antibodies and the cognate antigens are present. Finally, we describe a clinical case in which anti-CD177 present in a transfusion recipient precipitated TRALI after the transfusion of CD177-positive, but not CD177-negative, PRBCs. In conclusion, we introduce a new TRALI mechanism based on the specific binding of transfused, soluble antigens to activated ECs in preimmunized recipients. We suggest that further studies and clinical work-up of TRALI should also include antibody investigation of the recipient.
Introduction
Transfusion-related acute lung injury (TRALI) is a syndrome of respiratory distress precipitating in a timely context with the transfusion of blood components. Two major groups of elicitors are described in the literature: antibodies present in the blood component (antibody-mediated TRALI) and biological material that stems from blood storage lesions (non–antibody-mediated TRALI). TRALI is an underestimated and still underreported consequence of blood transfusion, and its definition has been clarified recently.1 Approximately 50% to 80% of TRALI cases have been related to the transfusion of antibodies against human neutrophil antigens (HNA) or human leukocyte antigens (HLA), mostly present in blood components produced from female donors with previous pregnancies.2 However, this percentage may be even higher, depending on the methods used.3 The implementation of male-only plasma measures has reduced the number of TRALI cases by approximately two-thirds.4,5 Even with this successful measure, TRALI remains a critical side effect of transfusion and is still a relevant cause of transfusion-related mortality in industrialized countries.6 Non-antibody elicitors may be responsible for the majority of remaining cases.1,7,8
Different animal models have proved the effects of lipid from stored blood, stored platelet-derived vascular endothelial growth factors, acid sphingomyelinase from aged platelets, microparticles, and accumulated soluble CD40 ligand in mechanisms of non–antibody-mediated TRALI. However, supporting data obtained from human cases are limited; in particular, red blood cell (RBC) storage time, which was related to TRALI in animal models, did not lead to significant effects in humans.1,6,9,10 Bioactive lipids produced contradictory results in animal studies and in clinical trials.11-14 No definite conclusion can be drawn from contradictory studies on the role of soluble CD40L.15-17
Reverse TRALI, a condition in which the transfusion recipient has preformed antibodies against an antigen present in the transfusion, has not been investigated in much depth after general blood filtration was introduced. In fact, “classical” reverse TRALI has not been reported in countries using universal leukodepletion, other than following the transfusion of (unfiltered) granulocyte concentrates.18,19 Investigating an unusual case of TRALI prompted us to question whether proteins released from donor cells during processing or storage of blood components would be capable of inducing a new type of reverse TRALI. Filtration will only take out white blood cells but not released proteins. Specific interactions between certain white blood cell proteins and their receptors on endothelial cells are well described. Here, we show that soluble neutrophil surface protein CD177 in complex with proteinase 3 (sCD177/PR3) is present in packed red blood cell (PRBC) supernatant, binds specifically to endothelium, and becomes recognized by preexisting anti-CD177 isoantibodies in the blood recipient’s circulation, which leads to a new type of reverse TRALI.
Materials and methods
Antibodies and proteins
The following monoclonal antibodies (mAbs) were used for this study: 7D8 against CD177 was produced in the Giessen laboratory (Institute for Clinical Immunology and Transfusion Medicine, Justus Liebig University, Giessen, Germany) from hybridomas, a generous gift from David Stroncek, National Institutes of Health (Bethesda, MD). Hybridoma clone-producing Gi18 was developed in our institute. MEM166 against CD177 (Serotec, Düsseldorf, Germany), PR3G-2 against PR3 (Santa Cruz Biotechnology, Dallas, TX), PR3-D1 (My BioSource, San Diego, CA), and monoclonal immunoglobulin G (mIgG) were purchased (Ancell, Bayport, NY). PECAM1.1 and PECAM1.2 were generous gifts from Peter Newman, Blood Research Institute (Milwaukee, WI). Antibodies against FCGRI (clone 10.1; BioLegend, San Diego, CA), FCGRII (clones IV.3 and AT10; BioLegend), and FCGRIIIB (3G8; Sigma-Aldrich, Darmstadt, Germany) were purchased.
Human anti–HNA-2 (n = 4) was obtained from serum from women who gave birth to children with neonatal alloimmune neutropenia. HNA-2 antibody specificity was identified by use of granulocyte immunofluorescence testing and mAb immobilization of granulocyte antigen assay. Sera containing anti-PR3 (n = 4) were obtained from patients with granulomatosis with polyangiitis, formerly known as Wegener’s granulomatosis. Control sera (n = 4) were collected from nonimmunized healthy blood donors. Use of human material was approved by the local ethics committee (Region North Jutland, Denmark, N-20170026; Justus Liebig University, Giessen, Germany, medical faculty no. 05/00). The study was performed in accordance with the Declaration of Helsinki. Native CD177/PR3 (nCD177/PR3) was isolated from neutrophil lysates as previously described.20 Recombinant CD177 (rCD177) were produced in our laboratory using transfected High Five cells (Invitrogen, Carlsbad, CA).21,22 Recombinant proteinase 3 (rPR3) was a generous gift from Triantafyllos Chavakis, University Hospital Carl Gustav Carus, Dresden, Germany, and PECAM-1 from Peter Newman, Blood Research Institute. Native PR3 was purchased from Fitzgerald Industries International (Acton, MA).
Isolation of IgG from human serum, generation of F(ab′)2 fragments
Total IgG from 1 mL of serum was purified with a Melon-Gel IgG Spin Purification Kit (Thermo Fisher Scientific, Rockford, IL) and dialyzed overnight at 4°C in phosphate-buffered saline (PBS) using Slide-A-Lyzer Dialysis Cassettes (10c000 molecular weight cut-off; Thermo Fisher Scientific). The protein concentration was measured by using the NanoDrop system (Thermo Fisher Scientific) and adjusted to a concentration of 60 mg/mL. To generate F(ab′)2 fragments, aliquots of 2 mg mAbs or 4 mg total human IgG (10 mg/mL) were digested with immobilized pepsin protease as recommended by the kit’s manufacturer (Thermo Fisher Scientific). Isolated F(ab′)2 fragments were dialyzed and measured by bicinchoninic assay (Thermo Fisher Scientific). F(ab′)2 fragments were analyzed on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis under reducing and nonreducing conditions and visualized by silver staining. Only proteins with purity >95% were used for further experiments.
Antibody deglycosylation
mAbs IV.3, AT10, 10.1, and 3G8 were deglycosylated by in vitro digestion with Endo S (New England Biolabs, Ipswich, MA).
Purification of sCD177/PR3
Plasma was isolated from full blood of CD177-positive donors. sCD177 was purified from plasma by using affinity chromatography using 1.5 mg mAb 7D8 coupled on a column (AminoLink, Thermo Fisher Scientific, Bonn, Germany), and purified protein was dialyzed in 20 mmol/L Tris-buffered saline buffer, pH 7.4. Protein concentration was determined by using bicinchoninic assay (Thermo Fisher Scientific).
Detection of sCD177/PR3 in enzyme-linked immunosorbent assay
For the detection of sCD177/PR3 in plasma, a sandwich enzyme-linked immunosorbent assay (ELISA) technique was established. Microtiter wells were coated with mAb MEM166 (5 µg) in buffer overnight at 4°C. After washings, 100 µL of human plasma was added. After 1 hour at 4°C, wells were washed with PBS/bovine serum albumin 0.02%, and either biotin-labeled mAb 7D8 or PR3-D1 (9 µg) was added. After incubation for 1 hour at room temperature, wells were washed again, 100 µL of horseradish peroxidase–labeled anti-human IgG (dilution 1:50c000; GE Healthcare, Chicago, IL) was added, and wells were incubated for 1 hour at room temperature. To detect bound antibodies, following a wash step, 3,3',5,5'-tetramethyl benzidine substrate solution (Sigma Healthcare, Rowville, VIC, Australia) was added; after 1 hour at room temperature, the reaction was stopped with 100 µL of 1.0 mol/L hydrochloric acid and read at 450 nm on an ELISA reader (Tecan, Männedorf, Switzerland). The cutoff values were determined by using the mean signal of the plasma from CD177-negative individuals (n = 7) plus 3 times standard deviations (SDs). In some experiments, supernatant from buffy coat–reduced RBC concentrates before and after in-line leukoreduction (Maco Pharma International, Langen, Germany) were collected. Collected supernatants were spun down at 155c000 g for 1 hour in an ultracentrifuge and subsequently analyzed. Albumin concentration was determined by using a human albumin ELISA kit (Abcam, Berlin, Germany).
Handling of endothelial cells
Primary human umbilical vein endothelial cells (HUVECs) were purchased from Lonza (Basel, Switzerland) and human pulmonary microvascular endothelial cells (HPMECs) were purchased from Creative Bioarray (New York, NY). Both cells were cultured in Lonza medium containing 25% fetal calf serum provided by the same company. HUVECs cultured for <4 passages were used in the experiments. For most experiments, cells (2 × 105) were cultured in 24-well plates and stimulated with PBS or 100-ng tumor necrosis factor-α (TNF-α) (ImmunoTools, Friesoythe, Germany). After overnight incubation, the medium was changed to serum-free medium (SFM); 2 µg/mL proteins of interest or saline were added for 4 hours at 37°C. For inhibition experiments, cells were pretreated with mAbs (1 µg) against PECAM-1 (Gi5, PEC1.1, or PEC1.2) for 2 hours at 37°C before the addition of proteins. HUVECs were detached with Accutase (PAA Laboratories GmbH, Cölbe, Germany), washed with PBS, and incubated with 0.5 µg fluorescein isothiocyanate (FITC)–labeled mIgG, MEM166, or PR3 D1 mAbs for 20 minutes in 4°C. After washings with PBS, cells were analyzed by using flow cytometry on an FACS Canto (BD Biosciences, Heidelberg, Germany).
For functional tests, after the addition of proteins, cells were exposed to 0.4 µg/200 µL MEM166, PR3 D1, or mIgG or 10 µL human serum overnight at 37°C before the cells were detached for analysis. Thrombin (0.2 U/mL; Chrono-Log Corporation, Havertown, PA) for 15 minutes, 37°C was used as a positive control.
Detection of FCGRs on endothelial cells
The expression of FCGRI, FCGRII, and FCGRIII on the surface of HPMECs was evaluated in ELISA. In brief, 4 × 105 cells were cultured in 96-well plates. The next day, the medium was removed, and cells were blocked by using 2% bovine serum albumin in PBS for 1 hour at 37°C. After washing with PBS, wells were incubated for 1 hour at 37°C with mAbs IV.3 or AT10 or 10.1 or 3G8 or mIgG (as isotype control). Antibodies against αv (P2W7; Santa Cruz Biotechnology, Heidelberg, Germany) integrin were used as positive control. After washing with PBS, cells were incubated with horseradish peroxidase–labeled anti-mouse IgG (dilution 1:50c000; GE Healthcare) for 1 hour at room temperature. Bound antibodies were detected with 3,3',5,5'-tetramethyl benzidine substrate solution (Sigma Healthcare). After 1 hour at room temperature, the reaction was stopped with 100 µL of 1.0 mol/L hydrochloric acid and read at 450 nm on an ELISA reader (Tecan).
Neutrophil isolation and testing
Neutrophils were isolated from EDTA-anticoagulated whole blood from healthy donors using dextran sedimentation. Neutrophils from phenotyped CD177-positive and CD177-negative donors (106) were incubated with cultured endothelial cells for 4 hours at 37°C. After washing with PBS, the membrane-bound CD177 or PR3 on endothelial cells were analyzed in flow cytometry.
sCD177/PR3 absorption to neutrophils
CD177-negative and CD177-positive phenotyped neutrophils (106) were incubated for 2 hours with 1 mL of serum from CD177-positive or CD177-negative donors at room temperature. Using FITC-labeled mAbs, the presence of CD177, PR3, and PECAM-1 on the neutrophil surface before and after incubation was evaluated by using flow cytometry. mIgG was used as isotype control.
sCD177/PR3 absorption from neutrophils to endothelial cells
HUVECs (2 × 105) were cultured in 24-well plates and stimulated with 100 ng of TNF-α. After overnight incubation, the medium was changed to SFM. Prestimulated HUVECs were incubated with neutrophils (106) from CD177-positive or CD177-negative individuals for 4 hours at 37°C. Using FITC-labeled mAbs, the presence of CD177 and PR3 on the endothelial surface after incubation with CD177-phenotyped neutrophils was analyzed.
Production of reactive oxygen species
The production of reactive oxygen species (ROS) in endothelial cells was measured by using 2′,7′-dichlorofluorescein diacetate (Abcam, Cambridge, UK) as previously described.23 In brief, HUVECs or HPMECs (2 × 105) were cultured in a 24-well plate overnight. The next day, medium was replaced with SFM containing 2′,7′-dichlorofluorescein diacetate (1.5 mL, 10 mmol/L) and subsequently stimulated as described earlier. Phorbol myristate (320 nmol/L; Sigma-Aldrich) was used as a positive control. The cells were detached and fixed in CELLFIX (250 µL, 1:10 dilution; Becton Dickinson, Heidelberg, Germany). ROS was measured by using flow cytometry. In some experiments before incubation with FITC-labeled mAbs, endothelial cells were treated with deglycosylated mAbs against FCGR (I, II, and III).
Permeability assay
The permeability through HUVEC monolayers was assessed by the passage of albumin-FITC (Sigma-Aldrich) as previously described.20 Briefly, HUVECs (2 × 105) were cultured for 2 days in transwell chambers with fibronectin-coated 3 µm pore-size polycarbonate membrane inserts (Costar, Cambridge, MA). For the assay, SFM containing albumin-FITC (100 µL, 40 mg/mL) was applied to the upper chamber. Endothelial monolayers were treated for 4 hours at 37°C with 2 µg/mL rCD177 or rPR3 or rCD177/PR3 followed by mAbs (2 µg/mL) against CD177 (MEM166) or PR3 (PR3 D1) as described earlier. In some experiments, before stimulation with protein, the endothelial monolayer was pretreated with PECAM1.2 F(ab′)2 fragments. The albumin-FITC concentration in the lower chamber was screened every 15 minutes over 1 hour in triplicate in a fluorescent microtiter plate reader (BioTek, Bad Friedrichshall, Germany). Thrombin (0.2 U/mL) was included as a positive control.
Apoptosis assay
HUVECs or HPMECs (1 × 105) were cultured on µ-slide wells (ibidi GmbH, Martinsried, Germany). The next day, cells were treated with proteins and antibodies as described earlier. As positive control, some cells were treated with RGD (40 µg/mL). Caspase-Glo 3/7 reagents (100 µL) were added to cells, and luminescence was measured by using a fluorescence microtiter reader (FLX800, BioTek Instruments, Winooski, VT).
Caspase activity was calculated by using luminescence values (of average 2 replicates from 3 independent experiments) relative to control.
Surface plasmon resonance analysis
Surface plasmon resonance (SPR) analysis was performed on a ProteOn XPR36 system (Bio-Rad, Hercules, CA) as previously described.20 PECAM-1 was immobilized onto the sensor chip (100 mg/mL) by amin coupling. mAbs against PECAM-1 (PEC1.2 or PEC 1.1) or rCD177, rPR3, and nCD177/PR3 at a concentration of 80 nM in PBS were injected as analytes over the chip at a flow rate of 20 and 100 mL/min, respectively, in a total volume of 250 mL at 25°C. The sensorgrams were then analyzed by using the ProteOn evaluation software package (Bio-Rad).
Effect of RBC filtration and storage on sCD177/PR3 concentration in the supernatant
Twelve units of PRBCs were obtained from the blood bank. Each unit was equally divided by weight into 2 parts. For 6 units, the first part was filtered by using standard blood bank technology. Supernatant was collected after ultracentrifugation directly or immediately after filtration. Non-leukoreduced blood (remaining 6 units) was stored at 4°C for 21 days, and supernatant before and after filtration was collected. Concentration of sCD177/PR3 before and after filtration in fresh and stored units was determined by ELISA.
Neutrophil/endothelial cell interaction
Neutrophils were isolated from EDTA-anticoagulated whole blood from healthy donors by using dextran sedimentation (Roth, Karlsruhe, Germany). Neutrophils (106) from CD177-positive and CD177-negative donors were incubated with cultured HUVECs for 4 hours at 37°C. After washings with PBS, membrane-bound CD177 or PR3 on HUVECs were analyzed by flow cytometry. In some experiments, isolated neutrophils (106) from CD177-negative phenotyped donors were incubated with serum (1 mL) from CD177-positive donors for 2 hours at room temperature. After washings with PBS, CD177 and PR3 were detected with specific mAbs by flow cytometry.
Statistical analysis
Data were analyzed by using Prism 8 (GraphPad Software, Inc., La Jolla, CA).
Results
sCD177 is present in human plasma
To prove the presence of sCD177 in human plasma, a sandwich ELISA was established. Donors were categorized, based on the percentage of CD177-positive neutrophils, as high (>70%), medium (between 30% and 70%), and low (<30%) or as CD177-negative. Plasma samples were added to mAb MEM166-coated wells, and immobilized sCD177/PR3 was detected by using biotin-labeled mAb 7D8. CD177 isolated from neutrophil lysates was used as positive control. No sCD177/PR3 was detectable in CD177-negative individuals (data not shown). The mean plasma concentration of sCD177/PR3 in CD177-phenotyped individuals was 0.62 ± 0.14 µg/mL, 0.23 ± 0.77 µg/mL, and 0.043 ± 0.01 µg/mL for high, medium, and low, respectively (Figure 1A). Because CD177 is expressed in complex with PR3 on the neutrophil surface, we questioned whether sCD177 would also carry PR3. Indeed, PR3 was detected after immobilization of sCD177 by the use of mAb PR3-D1 in the same ELISA system, indicating that both proteins together were captured on the plate (data not shown). To further support these initial findings, sCD177/PR3 complex was purified from plasma by affinity chromatography with MEM166. As shown in Figure 1B, under nonreducing conditions, purified sCD177 and nCD177 showed one band with an apparent molecular mass of 55 kDa and an additional band at 29 kDa. Immunoblotting confirmed those bands as CD177 and PR3, respectively.
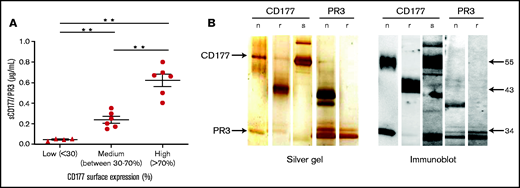
sCD177 (sCD177/PR3) is present in human plasma. (A) The percentage of CD177-positive neutrophils in healthy donors was assessed by using flow cytometry, and the amount of sCD177/PR3 in serum was measured by using ELISA. There is a tendency toward higher amounts of sCD177/PR3 in serum from individuals with a high proportion of CD177-positive neutrophils. **P < .005 (B) Analysis of affinity-purified sCD177/PR3 by silver staining (left panel) and immune blotting (right panel). nCD177, rCD177, “native” PR3 isolated from neutrophils (nPR3), and rPR3 were used for comparison. Blotted proteins were stained with anti-CD177 (human IgG) or anti-PR3 (PR3D1) mAbs. Bound antibodies were visualized by using a chemiluminescence system.
sCD177 (sCD177/PR3) is present in human plasma. (A) The percentage of CD177-positive neutrophils in healthy donors was assessed by using flow cytometry, and the amount of sCD177/PR3 in serum was measured by using ELISA. There is a tendency toward higher amounts of sCD177/PR3 in serum from individuals with a high proportion of CD177-positive neutrophils. **P < .005 (B) Analysis of affinity-purified sCD177/PR3 by silver staining (left panel) and immune blotting (right panel). nCD177, rCD177, “native” PR3 isolated from neutrophils (nPR3), and rPR3 were used for comparison. Blotted proteins were stained with anti-CD177 (human IgG) or anti-PR3 (PR3D1) mAbs. Bound antibodies were visualized by using a chemiluminescence system.
Endothelial cells specifically bind sCD177/PR3 via an interaction between PECAM-1 and PR3
We have previously described that CD177 on neutrophils is a heterophilic binding partner for endothelial PECAM-1, an interaction that is involved in neutrophil transmigration. Incubation of PBS-pretreated HUVECs with rCD177, rPR3, and sCD177/PR3 did not consistently result in binding of these proteins to endothelial cells as measured by flow cytometry (supplemental Figure 1A). In contrast, when HUVECs were pretreated with TNF-α, both rPR3 and sCD177/PR3 could be detected on the cell surface, whereas rCD177 was not absorbed (Figure 2A; supplemental Figure 1B). Blocking experiments with specific mAbs against domains 1, 2, and 6 of PECAM-1 revealed that the specific absorption of sCD177/PR3 and PR3 to endothelial cells is mediated via domain 6 of PECAM-1 (Figure 2B; supplemental Figure 1C), showing that CD177 and PECAM-1 interact via PR3, but not directly, as heterophilic binding partners.
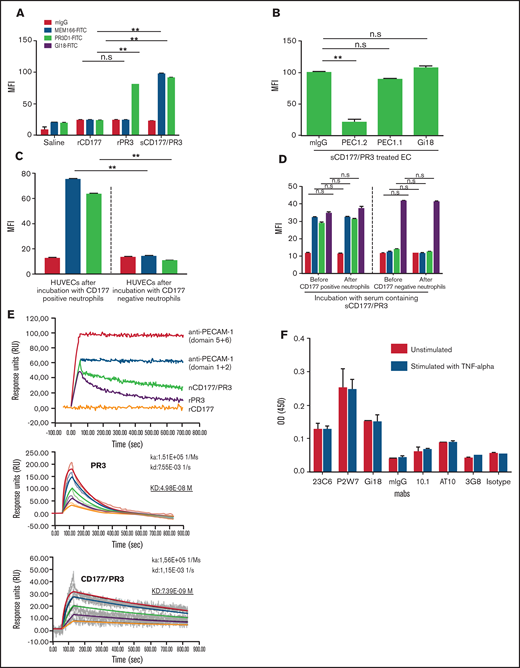
Endothelial cells (ECs) bind sCD177/PR3 specifically via PECAM-1. (A) HUVECs were pretreated with TNF-α overnight and incubated with saline, rCD177, rPR3, or sCD177/PR3 (2 µg/mL) for 4 hours at 37°C. HUVECs were analyzed by flow cytometry using direct fluorescence-labeled mAbs against CD177 (MEM166) or PR3 (PR3D1). Values are presented as mean fluorescence intensity (MFI) ± standard deviation (SD) (n = 5). (B) TNF-α–pretreated HUVECs were treated with mAbs (1 µg/mL), against PECAM-1 domain 1 (Gi18) or against PECAM domains 2 and 6 (PECAM 1.1 or PECAM 1.2, respectively), and subsequently incubated with sCD177/PR3 (2 µg/mL) for 4 hours at 37°C. HUVECs were analyzed by flow cytometry using direct fluorescence-labeled anti-CD177 (MEM166). Values are presented as MFI ± SD (n = 5). (C) Presence of CD177/PR3 and ECs after incubation with granulocytes. Neutrophils were isolated from whole blood of CD177-positive and CD177-negative phenotyped donors. HUVECs were incubated with granulocytes for 4 hours in 37°C. Presence of CD177 or PR3 on ECs was determined by flow cytometry. Bar graphs represent the MFI on the EC membrane. Values are presented as mean ± SD (n = 5). (D) Presence of CD177/PR3 on neutrophils after incubation with serum. CD177-negative and CD177-positive phenotyped neutrophils were isolated and incubated for 2 hours with 1 mL of serum from CD177-positive donors. Presence of CD177, PR3, and PECAM-1 on the neutrophil surface before and after incubation was determined by flow cytometry. Bar graphs represent the MFI on the neutrophil surface. Values are presented as mean ± SD (n = 5). (E) Binding of rCD177, rPR3, and sCD177/PR3 on coated PECAM-1 was determined in real-time by using SPR. Purified PECAM-1 was immobilized on a sensor chip. mAbs against PECAM-1 Ig domains 5 + 6 or 1 + 2 or rCD177, rPR3, or sCD177/PR3 were injected at 25°C. Note that rCD177 does not bind to PECAM-1 (upper panel). To calculate the KD for PR3 or sCD177/PR3 interaction with PECAM-1, different concentrations of both proteins were run as analyte on coated PECAM-1 (middle and lower panel). (F) ECs express FCGRI and FCGRII on the surface. The expression of FCGRs (I, II, and IIIb) on HPMEC monolayer were evaluated in ELISA. Endothelial monolayers were cultured in a 96-well plate and blocked with bovine serum albumin. mAbs against FCGRs (10.1 for FCGRI, AT10 for FCGRII, and 3G8 for FCGRIII) were added to ECs, and bound antibodies were detected by using horseradish peroxidase–labeled anti-mouse secondary antibodies. The optical densities (ODs) were evaluated in photometer.**P< .01, n.s., not significant.
Endothelial cells (ECs) bind sCD177/PR3 specifically via PECAM-1. (A) HUVECs were pretreated with TNF-α overnight and incubated with saline, rCD177, rPR3, or sCD177/PR3 (2 µg/mL) for 4 hours at 37°C. HUVECs were analyzed by flow cytometry using direct fluorescence-labeled mAbs against CD177 (MEM166) or PR3 (PR3D1). Values are presented as mean fluorescence intensity (MFI) ± standard deviation (SD) (n = 5). (B) TNF-α–pretreated HUVECs were treated with mAbs (1 µg/mL), against PECAM-1 domain 1 (Gi18) or against PECAM domains 2 and 6 (PECAM 1.1 or PECAM 1.2, respectively), and subsequently incubated with sCD177/PR3 (2 µg/mL) for 4 hours at 37°C. HUVECs were analyzed by flow cytometry using direct fluorescence-labeled anti-CD177 (MEM166). Values are presented as MFI ± SD (n = 5). (C) Presence of CD177/PR3 and ECs after incubation with granulocytes. Neutrophils were isolated from whole blood of CD177-positive and CD177-negative phenotyped donors. HUVECs were incubated with granulocytes for 4 hours in 37°C. Presence of CD177 or PR3 on ECs was determined by flow cytometry. Bar graphs represent the MFI on the EC membrane. Values are presented as mean ± SD (n = 5). (D) Presence of CD177/PR3 on neutrophils after incubation with serum. CD177-negative and CD177-positive phenotyped neutrophils were isolated and incubated for 2 hours with 1 mL of serum from CD177-positive donors. Presence of CD177, PR3, and PECAM-1 on the neutrophil surface before and after incubation was determined by flow cytometry. Bar graphs represent the MFI on the neutrophil surface. Values are presented as mean ± SD (n = 5). (E) Binding of rCD177, rPR3, and sCD177/PR3 on coated PECAM-1 was determined in real-time by using SPR. Purified PECAM-1 was immobilized on a sensor chip. mAbs against PECAM-1 Ig domains 5 + 6 or 1 + 2 or rCD177, rPR3, or sCD177/PR3 were injected at 25°C. Note that rCD177 does not bind to PECAM-1 (upper panel). To calculate the KD for PR3 or sCD177/PR3 interaction with PECAM-1, different concentrations of both proteins were run as analyte on coated PECAM-1 (middle and lower panel). (F) ECs express FCGRI and FCGRII on the surface. The expression of FCGRs (I, II, and IIIb) on HPMEC monolayer were evaluated in ELISA. Endothelial monolayers were cultured in a 96-well plate and blocked with bovine serum albumin. mAbs against FCGRs (10.1 for FCGRI, AT10 for FCGRII, and 3G8 for FCGRIII) were added to ECs, and bound antibodies were detected by using horseradish peroxidase–labeled anti-mouse secondary antibodies. The optical densities (ODs) were evaluated in photometer.**P< .01, n.s., not significant.
The same phenotype change of TNF-α primed endothelial cells was also observed after incubation with CD177-positive, but not CD177-negative, neutrophils (Figure 2C). In contrast, CD177-negative neutrophils, incubated with plasma containing sCD177/PR3, did not change their phenotype, despite the fact that they constitutively express PECAM-1 (Figure 2D).
We further corroborated this finding by SPR technology with immobilized PECAM-1 (Figure 2E). Both PR3 alone and in complex with CD177 showed specific binding, whereas no interaction was observed for rCD177. Interestingly, compared with PR3 alone, the complex revealed a more stable interaction. Dilution studies revealed a KD of 7.39E-09 M for CD177/PR3 complex vs a KD of 4.98E-08 M for PR3 alone.
Endothelial cells express FCGRI and FCGRIIa
To clarify the potential role of FCGRs in the mechanism of endothelial dysfunction induced by binding of antibodies against CD177 or PR3 on absorbed sCD177/PR3, the presence of FCGRs on the endothelial cell monolayer was investigated. FCGRIIIb was not detectable. Compared with the expression of PECAM-1 or αv integrin, FCGRII exhibited weak, and FCGRI exhibited very weak, expression on the endothelial surface. The expression was independent of TNF-α stimulation (Figure 2F).
Antibodies against absorbed sCD177/PR3 induce endothelial cell dysfunction
We have previously reported that the interaction between CD177/PR3 on neutrophils and PECAM-1 on endothelial cells can induce PECAM-1–mediated signaling, leading to VE-cadherin degradation and endothelial barrier instability.20 The consequence of cross-linking sCD177/PR3 bound to PECAM-1 with specific antibodies was therefore subsequently studied.
First, ROS production in the presence of anti-PR3 and anti-CD177 after preincubation of TNF-α–pretreated endothelial cells with rCD177, rPR3, or sCD177/PR3 was assessed (Figure 3A). A monoclonal anti-CD177 induced ROS production only in cells preincubated with sCD177/PR3 (mean fluorescence intensity 4.3-fold over control) but not rCD177 or rPR3. IgG fractions prepared from sera from anti-CD177 immunized patients induced comparable effects. In addition, binding of monoclonal anti-PR3 induced ROS production in HUVECs after preincubation with sCD177/PR3 or rPR3 (mean fluorescence intensity 2.5-fold and 3-fold over control, respectively). In a similar set of experiments (Figure 3B, upper panel), incubation of HUVECs with F(ab′)2 fragments induced ROS production with an identical pattern but with an ∼25% decrease in the overall response, indicating a partially FcR-dependent mechanism.

Antibodies against sCD177/PR3 have a functional impact on endothelial cells. (A) To evaluate the production of ROS, TNF-α–pretreated HUVECs or HPMECs were incubated with rCD177 (open bars), rPR3 (gray bars), or sCD177/PR3 (black bars), followed by antibodies or controls as indicated. Phorbol myristate (PMA) was used as assay control. ROS was measured by flow cytometry using 2',7'-dichlorofluorescein diacetate as a fluorochrome. Values are presented as mean fluorescence intensity (MFI) ± standard deviation (n = 5). (B) Experiments were performed as in panel A using F(ab′)2 fragments instead of complete antibodies (upper panel) or pretreated with deglycosylated anti-FCGR antibodies (I, II, and IIIb) before antibody binding (lower panel). (C) To evaluate the induction of apoptosis, endothelial cells were incubated with rCD177 (open bars), rPR3 (gray bars), or sCD177/PR3 (black bars), followed by antibodies or controls as indicated. HPMECs (dotted bars) were incubated with sCD177/PR3 followed by antibodies or controls as indicated. RGD (40 mg/mL) was used as assay control. Caspase-3/7 activity was then measured. Values are presented as mean ± standard deviation (n = 5). (D) To evaluate endothelial permeability, HUVECs (upper panel) or HPMECs (lower panel) were cultured on fibronectin-coated polycarbonate filter chambers for 48 hours and treated with rCD177 (open bars), rPR3 (gray bars), or sCD177/PR3 (black bars), followed by incubation with mAbs or controls as indicated. Thrombin (0.2 U/mL) was used as an assay control. The passage of FITC-albumin through the monolayer of cells was measured at different time points (5–60 minutes). Values are presented as mean ± standard deviation (n = 5). (E) Experiments were performed as in panel D in the presence of mAb PECAM1.2 F(ab′)2 fragments. d, deglycosylated; *P< .05, **P < .01, ***P < .001, n.s. not significant.
Antibodies against sCD177/PR3 have a functional impact on endothelial cells. (A) To evaluate the production of ROS, TNF-α–pretreated HUVECs or HPMECs were incubated with rCD177 (open bars), rPR3 (gray bars), or sCD177/PR3 (black bars), followed by antibodies or controls as indicated. Phorbol myristate (PMA) was used as assay control. ROS was measured by flow cytometry using 2',7'-dichlorofluorescein diacetate as a fluorochrome. Values are presented as mean fluorescence intensity (MFI) ± standard deviation (n = 5). (B) Experiments were performed as in panel A using F(ab′)2 fragments instead of complete antibodies (upper panel) or pretreated with deglycosylated anti-FCGR antibodies (I, II, and IIIb) before antibody binding (lower panel). (C) To evaluate the induction of apoptosis, endothelial cells were incubated with rCD177 (open bars), rPR3 (gray bars), or sCD177/PR3 (black bars), followed by antibodies or controls as indicated. HPMECs (dotted bars) were incubated with sCD177/PR3 followed by antibodies or controls as indicated. RGD (40 mg/mL) was used as assay control. Caspase-3/7 activity was then measured. Values are presented as mean ± standard deviation (n = 5). (D) To evaluate endothelial permeability, HUVECs (upper panel) or HPMECs (lower panel) were cultured on fibronectin-coated polycarbonate filter chambers for 48 hours and treated with rCD177 (open bars), rPR3 (gray bars), or sCD177/PR3 (black bars), followed by incubation with mAbs or controls as indicated. Thrombin (0.2 U/mL) was used as an assay control. The passage of FITC-albumin through the monolayer of cells was measured at different time points (5–60 minutes). Values are presented as mean ± standard deviation (n = 5). (E) Experiments were performed as in panel D in the presence of mAb PECAM1.2 F(ab′)2 fragments. d, deglycosylated; *P< .05, **P < .01, ***P < .001, n.s. not significant.
Similarly, incubation of HPMECs with deglycosylated anti-FCGRIIa or anti-FCGRI before endothelial treatment with MEM166 decreased ROS production up to 19% and 14%, respectively. No influence of FCGRIIIb blockage on endothelial ROS production was observed (Figure 3B, lower panel).
Second, endothelial cell apoptosis in the presence of anti-PR3 and anti-CD177 before and after preincubation with rCD177, rPR3, or sCD177/PR3 was assessed (Figure 3C). The observed pattern of apoptosis induction was fully consistent with our data obtained in the ROS assay: mAbs against CD177 and human anti-CD177 IgG induced apoptosis in endothelial cells preincubated with sCD177/PR3 only. Similarly, an mAb against PR3 in HUVECs preincubated with PR3 alone or with sCD177/PR3 induced apoptosis, indicating that the binding of PR3 to PECAM-1, alone or in complex with sCD177, provides a signaling complex that in presence of anti-PR3 or anti-CD177 antibodies can trigger apoptosis in endothelial cells. Comparable to HUVECs, HPMECs preincubated with sCD177/PR3 also undergo apoptosis after binding of mAb against CD177, human anti-CD177 IgG, or mAb against PR3.
Third, to assess the potential consequences of signals triggered in endothelial cells in the presence of antibodies (anti-PR3 or anti-CD177) upon binding of sCD177/PR3 or PR3, a permeability assay was performed (Figure 3D). HUVECs (upper panel) or HPMECs (lower panel) were cultured in transwell chambers up to confluency and preincubated with rCD177, rPR3, or sCD177/PR3 for 4 hours, followed by the addition of saline or mIgG, MEM166, PR3-D1, or human anti-CD177 IgG. The influx of FITC-labeled albumin through the endothelial monolayer toward the lower chamber was analyzed. The reaction pattern was comparable to our observations from the ROS production and apoptosis studies: in HUVECs preincubated with rPR3, only mAb PR3-D1 enhanced permeability (3.8 ± 0.2 fold; P < .0001). In HUVECs preincubated with sCD177/PR3, an increased permeability was induced by antibodies against CD177 (MEM166, 4.1 ± 0.6-fold; human IgG, 3.7 ± 0.5-fold) and antibodies against PR3 (3.6- ± 0.4-fold). No barrier disturbance could be induced with mIgG or any of the antibodies after preincubation with rCD177.
All effects were abrogated when endothelial cells were pretreated with F(ab′)2 fragments of mAb PECAM1.2 before incubation with rPR3 or sCD177/PR3 (Figure 3E).
RBC filtration and storage induces CD177/PR3 shedding from neutrophils
Having shown the presence of CD177/PR3 complexes in plasma, we next questioned whether almost plasma-free blood components, namely, PRBCs, may also contain relevant amounts of sCD177/PR3. As shown in Figure 4A, a 3.7-fold mean increase in the concentration of sCD177/PR3 was detected after filtration. In contrast, albumin concentration did not differ before or after filtration.

sCD177/PR3 is detectable in the supernatant from RBC concentrates. (A) The concentration of sCD177/PR3 was measured in fresh RBCs (day 1) and stored RBCs (day 21) before and after filtration using standard blood bank procedures (left). As control, the albumin concentration in RBCs before and after filtration was assessed (right). (B) Evaluation of sCD177/PR3 protein content in sera from TRALI patients and 3 donors. The serum sample from the TRALI patient and 3 donors were analyzed in ELISA. Note, donor 1 was involved in TRALI reaction. The values are representative of 3 independent tests and are presented as mean ± SD. **P < .01.
sCD177/PR3 is detectable in the supernatant from RBC concentrates. (A) The concentration of sCD177/PR3 was measured in fresh RBCs (day 1) and stored RBCs (day 21) before and after filtration using standard blood bank procedures (left). As control, the albumin concentration in RBCs before and after filtration was assessed (right). (B) Evaluation of sCD177/PR3 protein content in sera from TRALI patients and 3 donors. The serum sample from the TRALI patient and 3 donors were analyzed in ELISA. Note, donor 1 was involved in TRALI reaction. The values are representative of 3 independent tests and are presented as mean ± SD. **P < .01.
In parallel experiments, the sCD177/PR3 concentration in unfiltered and filtered PRBC after 3 weeks of storage was evaluated. Storage induced a 5.5-fold mean increase in sCD177/PR3 concentration (Figure 4A). Only a slight increase in sCD177/PR3 concentration was observed after storage of filtered PRBCs.
TRALI after the transfusion of a leukodepleted unit of RBCs
A 40-year-old female patient was diagnosed with colorectal carcinoma and underwent surgery. After the surgical procedure, she required PRBC transfusions because of perioperative bleeding. After the first 120 mL of leuko-reduced PRBC, she developed severe acute respiratory distress with tachypnea (40 breaths per minute, oxygen saturation 70% on room air), tachycardia (145 beats per minute), and a sudden copious frothy tracheal exudate. An echocardiography revealed normal left ventricular function.
The patient was intubated and mechanically ventilated. Diuretics were given without effect. Her chest radiographs showed bilateral diffuse infiltrates consistent with acute TRALI. She was phenotyped as CD177-negative, and the serologic work-up revealed the presence of anti-CD177 isoantibodies in her plasma (developed during previous pregnancy), whereas no antibodies were detected in the donor. Due to the leukemia, the patient was severely anemic (hemoglobin 4.5 mmol/L before transfusion). The anti-CD177 antibody had a titer of 1:4096. Two units of leuko-reduced RBCs from CD177-negative donors were later transfused, without side effects. Analysis of supernatant from transfused PRBCs showed presence of sCD177/PR3 in the implicated PRBCs only; no sCD177/PR3 was detected in the PRBCs from non–TRALI-inducing donors (Figure 4B).
Discussion
In this study, we show the presence of sCD177/PR3 in plasma and PRBC storage solution. Our functional data elucidate that sCD177/PR3 can bind to PECAM-1 on stimulated endothelial cells, and that exposure of sCD177/PR3-positive endothelial cells to antibodies against CD177 subsequently induces endothelial activation and severe barrier dysfunction. This mechanism resembles in vitro changes observed in TRALI.
In line with the proposed two-hit or threshold model of TRALI, the mechanism induced by sCD177/PR3 requires endothelial cell activation as the “first hit.”24,25
We pretreated endothelial cells with TNF-α, which is known as a key cytokine in various pulmonary diseases such as acute lung injury.26 Whereas the analysis of cytokine profiles in TRALI patients before and after the reaction showed no difference in TNF-α concentrations,27 TNF-α levels were significantly elevated in TRALI patients compared with transfused patients without TRALI in a more recent study.28 It has long been known that endothelial exposure to cytokines induces the redistribution of PECAM-1, away from endothelial junctions, which, in our model system, may facilitate its approachability for ligands, including sCD177/PR3.29 Furthermore, antibody-binding studies have shown that TNF-α (but not interleukin-1β) induces conformational changes of PECAM-1.30 These changes probably also alter the accessibility of PECAM-1 for sCD177/PR3.
In contrast to other TRALI models, the “second hit” that we propose here is binding of sCD177/PR3 and its cognate antibody to PECAM-1. We have observed that sCD177/PR3 present in plasma and sCD177/PR3 shed from neutrophils are both capable of binding to endothelial cells; in addition, this mechanism is mediated via PECAM-1, since a specific mAb against heterophilic domains of PECAM-1 abrogates the binding completely. The interaction between endothelial PECAM-1 and neutrophil-derived CD177 was already reported several years ago.31 Our new data confirm that PR3 acts as the counter-receptor for PECAM-1 but also that the binding to PECAM-1 is strengthened when PR3 is in complex with CD177.32
The binding of anti-CD177 antibodies to sCD177/PR3 absorbed to PECAM-1 is required to initiate barrier breakdown in our model, most likely by clustering PECAM-1 and subsequent downstream signaling. In vivo, it is possible that sCD177/PR3 present in the transfused component will first form an immune complex with anti-CD177 in circulation, and then bind to endothelial PECAM-1 in the lung. We did not observe a difference in albumin flux through endothelial monolayers when sCD177/PR3 was preincubated with anti-CD177 (data not shown).
In response to cytokine stimulation, endothelial cells also upregulate Fc-γ receptor expression.33 Previous studies have documented the expression of FCGRIIa on the endothelial surface.33,34 Stimulation of aortic endothelial cells with TNF-α and interferon-γ in a dose-dependent manner significantly enhanced the expression of FCGRIIa.33 Immune complex–mediated cross-linking of FCGRIIa on endothelial cells triggers increased calcium influx and leads to endothelial activation.35,36
Our analysis detected a weak expression of FCGRIIa and very weak expression of FCGRI on the endothelial surface. These expressions were nonresponsive to TNF-α stimulation. The simultaneous binding of an antibody’s Fc part has been shown to induce cumulative effects on signaling initiated by the binding of F(ab′)2 fragments to the receptor.37 In line with this observation, binding of total human IgG to sCD177/PR3 absorbed on endothelial cells showed increased effects compared with F(ab′)2 fragments. Blockage of FCGRI and FCGRIIa, using deglycosylated mAbs before antibody binding on absorbed sCD177/PR3 on endothelial cells, limited the release of ROS significantly. Similar observations for the involvement of FCGRs in endothelial cell activation were reported.35
As a result of receptor clustering and signaling, endothelial cells produce ROS,23 which leads to cytokine production, enhanced cascade of polymorphonuclear leukocyte–endothelial interactions, and finally endothelial dysfunction (eg, apoptosis, permeability).38 Notably, a gp91phox knockout mouse was protected from antibody-mediated TRALI.39
At the end of the intracellular signaling cascade, endothelial barrier breakdown leads to a significant shift of albumin through the monolayer; this functional effect was seen with both HUVECs and HPMECs and again was completely abolished when the heterophilic domains of PECAM-1 were blocked.
All endothelial processes could be observed in the absence of neutrophils, supporting the hypotheses that neutrophils are not always required to precipitate TRALI.40,41 We are currently unable to answer the question of why CD177-negative neutrophils, which constitutively express PECAM-1, are unable to bind sCD177/PR3; apparently, TNF-α–mediated changes in PECAM-1 accessibility are specific to endothelial PECAM-1.
Finally, we have shown here that sCD177/PR3 is present in packed RBC concentrates. Leukoreduction by filtration has been shown to be effective in preventing relevant adverse reactions of transfusion, and most likely also TRALI, because leukocytes are responsible for some of the changes that occur in a stored unit during its shelf life.42 However, filtration is also known to activate neutrophils, leading to the release of granule contents in supernatant.43 Similar to this observation, our analysis of PRBC supernatants showed an increase of sCD177/PR3 after filtration.
Our results suggest that the recipient’s antibody status could play a more significant role in understanding TRALI than previously anticipated. Some TRALI cases assumed to be “non-antibody”–mediated TRALI probably follow such a mechanism. Only recently, De Clippel et al44 reported on high numbers of what they assumed to be “reverse TRALI” due to anti-HLA identified in the recipient. Similarly to the mechanism unraveled here, soluble HLA class I proteins in plasma and a yet-to-be-defined endothelial binding partner could have exerted these reactions.
However, in the absence of an animal model, the complete underlying mechanism of reverse TRALI is not yet fully understood. Appropriate in vivo studies will have to be conducted.
In summary, we introduce a new TRALI mechanism based on the absorption of transfused, soluble antigens to activated endothelial cells in preimmunized recipients. We suggest that further studies and clinical work-up of TRALI cases should also include antibody investigation of the recipient.
Acknowledgments
The authors are grateful to Christine Hoffmann and Heike Berghöfer for their excellent technical assistance.
Authorship
Contribution: B.B. and U.J.S. designed the study and conducted the experiments; K.R.N. provided the patients’ serum and clinical data of the TRALI case; A.T. conducted apoptosis experiments; M.B.-R. performed the SPR experiments; G.B. provided useful suggestions for the improvement of the experiments and reviewed the manuscript; B.B. analyzed and interpreted the data; and B.B. and U.J.S. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ulrich J. Sachs, Institute for Clinical Immunology and Transfusion Medicine, Justus Liebig University, Langhansstr 7, 35392 Giessen, Germany; e-mail: ulrich.sachs@med.uni-giessen.de.

