

Key Points
SCD-induced chronic hemolysis reduces lung HUWE1 and miR-98 through suppression of PPARγ and HUWE1.
Targeting PPARγ activation induces ubiquitination of NF-kB/p65 via HUWE1 signaling pathways in SCD-PH pathogenesis.

Abstract
Sickle cell disease (SCD)-associated pulmonary hypertension (PH) causes significant morbidity and mortality. Here, we defined the role of endothelial specific peroxisome proliferator-activated receptor γ (PPARγ) function and novel PPARγ/HUWE1/miR-98 signaling pathways in the pathogenesis of SCD-PH. PH and right ventricular hypertrophy (RVH) were increased in chimeric Townes humanized sickle cell (SS) mice with endothelial-targeted PPARγ knockout (SSePPARγKO) compared with chimeric littermate control (SSLitCon). Lung levels of PPARγ, HUWE1, and miR-98 were reduced in SSePPARγKO mice compared with SSLitCon mice, whereas SSePPARγKO lungs were characterized by increased levels of p65, ET-1, and VCAM1. Collectively, these findings indicate that loss of endothelial PPARγ is sufficient to increase ET-1 and VCAM1 that contribute to endothelial dysfunction and SCD-PH pathogenesis. Levels of HUWE1 and miR-98 were decreased, and p65 levels were increased in the lungs of SS mice in vivo and in hemin-treated human pulmonary artery endothelial cells (HPAECs) in vitro. Although silencing of p65 does not regulate HUWE1 levels, the loss of HUWE1 increased p65 levels in HPAECs. Overexpression of PPARγ attenuated hemin-induced reductions of HUWE1 and miR-98 and increases in p65 and endothelial dysfunction. Similarly, PPARγ activation attenuated baseline PH and RVH and increased HUWE1 and miR-98 in SS lungs. In vitro, hemin treatment reduced PPARγ, HUWE1, and miR-98 levels and increased p65 expression, HPAEC monocyte adhesion, and proliferation. These derangements were attenuated by pharmacological PPARγ activation. Targeting these signaling pathways can favorably modulate a spectrum of pathobiological responses in SCD-PH pathogenesis, highlighting novel therapeutic targets in SCD pulmonary vascular dysfunction and PH.
Introduction
Sickle cell disease (SCD) is an inherited red blood cell disorder.1,2 Millions of patients worldwide and ∼100 000 people in the United States suffer from SCD.3,4 Pulmonary hypertension (PH) is an increasingly recognized complication of SCD that is associated with high morbidity and mortality.5-7 The pathogenesis of PH complicating SCD involves endothelial dysfunction with increased production of vasoconstrictors, for example, endothelin-1 (ET-1),8-18 and binding of inflammatory cells to vascular endothelium.12 Previous reports indicate that 6% to 10.4% of patients with SCD have a mean pulmonary arterial pressure ≥25 mm Hg.8,19,20 Given the recent expanded definition of PH21,22 to include patients with mean pulmonary arterial pressure 21 to 25 mm Hg, these estimates may underestimate PH frequency in SCD. Furthermore, the pathogenic mechanisms underlying PH in SCD are poorly understood.
Transgenic sickle cell (SS) mice, including Berkeley SS mice,23,24 and the spherocytosis model25 spontaneously develop PH. Recently, we demonstrated for the first time that Townes humanized SS mice spontaneously develop vascular remodeling, and that hemin (HEM), which is released during hemolysis, reduces levels of the ligand-activated transcription factor, peroxisome proliferator-activated receptor γ (PPARγ), in SS mouse lungs and human pulmonary artery endothelial cells (HPAECs).11 As recently reviewed,26 current evidence indicates that reduced PPARγ is associated with PH pathogenesis,27-29 whereas PPARγ activation with thiazolidinedione ligands attenuates PH in experimental models.29-33 Our recent findings indicate that PPARγ activation favorably modulates levels of microRNAs (miRNAs or miR), including miR-27a through suppression of ETS Proto-Oncogene 1, Transcription Factor in SCD-PH.11 Collectively, these reports indicate that PPARγ activation provides a novel therapeutic approach in SCD-PH.
miRNAs, endogenous small noncoding RNAs of ∼22 nucleotides,34 are an important class of posttranscriptional regulators. miRNAs bind to the 3′ untranslated region of the targeted messenger RNA (mRNA) to negatively regulate gene expression. This binding stimulates translational repression or mRNA degradation of the target mRNA.34 miRNAs can impact cell cycle regulation, differentiation, apoptosis, and proliferation.35 Therefore, miRNAs are an important class of posttranscriptional regulators that are differentially expressed during development and disease. Rapidly emerging evidence demonstrates that dysregulation of miRNAs participates in PH pathogenesis36-41 and that miRNAs regulate pulmonary vascular cell remodeling and proliferation. However, the role of PPARγ-regulated miRNAs in endothelial cell biology in SCD-PH remains largely unknown. We recently elucidated that PPARγ loss of function decreases miR-98, leading to increases in ET-1 levels in hypoxia-induced PH.42 In contrast, PPARγ gain of function stimulates miR-98 levels and reduces ET-1.42
The current study further examines the regulation of miR-98 in SCD-PH. miR-98, which is located in intron 59 of its host gene, HUWE1,43 is positively associated with HUWE1 expression.44 HUWE1, a HECT (homology to E6-AP C terminus) E3 ubiquitin ligase, plays an important role in apoptosis, DNA replication, and DNA damage repair.34 HUWE1 also targets proteins for proteasomal degradation. Interestingly, in silico analysis of the HUWE1 promoter revealed putative PPARγ binding sites. We previously demonstrated that PPARγ loss of function increases NF-κB/p65 in hypoxia-induced PH,45 whereas PPARγ gain of function attenuates NF-κB levels.46 Furthermore, we and others reported that PPARγ negatively regulates NF-κB/p65 expression by a transrepression mechanism.45,47 A recent study reported that PPARγ induces ubiquitination and degradation of p65.48 These findings led us to postulate that chronic hemolysis in SCD stimulates reductions in PPARγ, loss of HUWE1 and miR-98, and increased p65 levels through reductions in its ubiquitination and degradation. These pathways promote NF-κB activation and endothelial dysfunction in SS mouse lungs and HEM-treated HPAECs. We further hypothesized that PPARγ activation attenuates these derangements, suggesting that therapeutic targeting of this receptor might provide a novel strategy for reducing SCD-associated pulmonary endothelial dysfunction and PH.
Materials and methods
Mouse models
As recently described, the Townes humanized mouse model of SCD (SS) was employed in the current study.11,49 ePPARγKO or ePPARγOX mice were generated as we reported.42 Chimeric mice were generated by transplanting SCD bone marrow from Townes mice using methods we previously published50-53 into littermate control (LitCon) or ePPARγKO mice as we have reported42,54 creating SSePPARγKO mice. Briefly, cohorts of 8- to 10-week-old recipient mice were subjected to lethal irradiation (6 cGy, twice, 4 hours apart) and transplanted with 2 × 107 bone marrow cells from homozygous sickle (SS) or control (AA) mice to generate chimeric AALitCon, AAePPARγKO, SSLitCon, and SSePPARγKO mice. LitCon or ePPARγKO mice were irradiated and transplanted with AA or SS bone marrow. Six-week post–bone marrow transplant, engraftment occurred, and the animals appeared healthy.
Measurement of PH in SS and chimeric mice
Selected male SS mice were gavaged with vehicle or the PPARγ ligand, rosiglitazone (RSG; 10 mg/kg per day) daily for 10 days as we have reported.11 SS, chimeric littermate control (AALitCon or SSLitCon), and chimeric (AAePPARγKO SSePPARγKO) mice at age 15 to 17 weeks were subjected to measurements of right ventricular systolic pressure (RVSP) and right ventricular hypertrophy (RVH) as reported.33 Furthermore, we examined PH of young (7 weeks) and late adult (26 weeks) mice because PH is the greatest risk factor for aged SCD patients.55 Briefly, after being anesthetized with isoflurane, RVSP was measured by introducing a 1.2F microtip pressure transducer catheter (Transonic) into the right internal jugular vein and advancing it to the right ventricle (RV), and RV pressures were continuously monitored for 10 minutes. RVH was assessed by dissecting the heart and determining the ratio of the weight of the RV to the weight of the left ventricle (LV) and septum (S) (Fulton’s index, RV: [LV + S] weight ratio). Selected animals were gavaged daily with RSG (10 mg/kg per day in 100-μL 0.5% methyl cellulose) or with an equivalent volume of vehicle for 10 days. All experiments using mice were approved by the Institutional Animal Care and Use Committee of the Atlanta Veterans Affairs Medical Center.
Reagents
HPAECs were obtained from ScienCell Research Laboratories (Carlsbad, CA). HUWE1, ET-1, PPARγ, and VCAM1 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The following reagents were obtained from Sigma-Aldrich (St Louis, MO): glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody, HEM, fetal bovine serum (FBS), and dimethyl sulfoxide (DMSO). BrdU proliferation assay was purchased from Abcam (Cambridge, MA). The PPARγ ligand, RSG, was obtained from Cayman Chemical (Ann Arbor, MI). Cell adhesion assay was purchased from Thermo Fisher Scientific (Waltham, MA).
HEM-treated HPAECs and functional assays
SCD and intravascular hemolysis expose the pulmonary endothelium to HEM activating adhesion molecule expression and increasing endothelial permeability and extravasation of fluid to the interstitial compartment.56 To model this stimulus, HPAECs were treated with pathophysiologically relevant HEM concentrations (5 μM) in vitro as we previously reported11 to further examine endothelial signaling pathways activated in SCD. Plasma heme concentrations in this range (∼4.2 μM compared with 0.2 μM in healthy controls [CON]) have been measured in patients with SCD.57 To examine the effect of RSG on HEM-induced cell proliferation and cell adhesion, HPAECs were treated with DMSO vehicle as a CON or with HEM (5 μM) dissolved in DMSO for 72 hours as reported.58 Selected HPAECs were also treated ± RSG (10 μM) or an equivalent volume of vehicle during the last 24 hours of HEM treatment, and cell proliferation was measured using BrdU proliferation assays as we have reported.46 Monocyte adhesion assays were performed according to the manufacturer’s instructions (Thermo Fisher Scientific). Briefly, THP1 monocytes (5 × 104 monocytes per well) labeled with calcein AM (5 µM) were added to confluent HPAEC monolayers pretreated with HEM (0 to 10 µM) and incubated for 30 minutes. Monolayers were then washed 3 times to remove unbound monocytes and scanned with a plate reader to measure total fluorescence per well.
miR-98 overexpression
To confirm the role of miR-98 in alterations in ET-1 expression, HPAECs (passages 3 to 6) were transfected with mimic-miR-98 (10 nM) or an equivalent amount of scrambled mimic-miR negative CON using lipofectamine RNAiMax (Qiagen) as we previous reported.42 After transfection for 6 hours, media were replaced with endothelial growth medium (EGM) containing 5% FBS. HPAECs were then treated with HEM (5 μM) for 72 hours. Alterations in miR-98 and ET-1 levels were examined using real-time quantitative polymerase chain reaction (qPCR).
PPARγ overexpression in HPAECs in vitro
To overexpress PPARγ, HPAECs were transfected with adenovirus containing a PPARγ plasmid (AdPPARγ, 0-50 multiplicity of infection) or CON GFP plasmid as we previously reported.42 Six hours after transfection, adenovirus-containing medium was replaced with fresh 5% FBS EGM, and HPAECs were then treated with DMSO or HEM (5 μM) for 72 hours. Levels of miR-98, HUWE1, ET-1, p65, PPARγ, and VCAM1 were determined by real-time qPCR.
HPAEC transfection and RNA interference studies
HPAECs were transfected with scrambled or HUWE1, p65 RNAi duplex (0 to 20 nM), or siPPARγ as we previously reported,42 using Lipofectamine 3000 Transfection Reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. After transfection for 6 hours, the transfection media was replaced with EGM containing 5% FBS. HPAECs were then treated with DMSO or HEM (5 μM) for 72 hours. HPAEC lysates were then harvested and examined for HUWE1, miR-98, p65, ET-1, VCAM1, and PPARγ levels using real-time qPCR and western blot assays.
p65 half-life and ubiquitination
To examine the half-life of p65, HPAECs were plated in 6-well plates (500 000 cells per well) and cultured in complete EGM containing 1% (v/v) penicillin/streptomycin. Twenty-four hours after plating, HPAECs were exposed to 40 to 100 μg/mL cycloheximide (CHX) in the presence or absence of 20 μM MG132 or 20 μM leupeptin for 0, 2, 4, or 8 hours. To determine if HUWE1 induces p65 ubiquitination, HPAECs were treated with HUWE1 plasmid (oxHUWE1, 1 to 2 μg) or vector (VEC) constructs for 6 hours and then incubated for an additional 72 hours. HPAEC lysates were then harvested and examined for HUWE1 and p65 levels using real-time qPCR or western blot assays. HUWE1 VEC was generously provided by Jingjing Wang (New York University, New York, NY). To further examine the effects of HUWE1 overexpression on HEM-treated HPAECs, after transfection with HUWE1 plasmids for 6 hours, HPAECs were then treated with DMSO or HEM (5 μM) for 72 hours. Levels of HUWE1, p65, ET-1, and VCAM1 were determined by real-time qPCR. HPAECs cells were cultured for 24 hours and then transfected with HA-ubiquitin plasmid (1 μg per well in a 6-well plate) using Lipofectamine 3000 according to the manufacturer’s recommendations. Twenty-four hours later, HPAEC lysates were collected in 1× M-PERTM Mammalian Protein Extraction Reagent from Thermo Fisher Scientific spiked with protease/phosphatase inhibitors examined by immunoblotting.
miRNA and mRNA real-time qPCR analysis
To measure miR-98, PPARγ, HUWE1, ET-1, p65, and VCAM1 levels in HPAECs or mouse lungs, small RNAs (<200 nt), and large RNAs (>200 nt) were isolated using the mirVana Kit (Invitrogen). The levels of miR-98 expression were analyzed by real-time qPCR using Qiagen miRNA Primer Assay according to the manufacturer’s instructions. RNU6B (miRNA) was used as an endogenous CON. PPARγ, HUWE1, ET-1, p65, and VCAM1 mRNA levels in the same sample were determined and quantified using specific mRNA primers as previously described.46 GAPDH or 9S mRNA levels were used as an endogenous CON.
Western blot analysis
After treatment with RSG or vehicle, protein homogenates from mouse lungs or HEM-treated HPAEC were subjected to western blot analysis as reported.46 Primary antibodies were purchased from Santa Cruz Biotechnology and included ET-1 rabbit polyclonal antibody (1:250 dilution, SC-98727, 24 kDa), NF-κB/p65 rabbit polyclonal antibody (1:500 dilution, SC-372, 65 kDa), PPARγ rabbit polyclonal antibody (1:500 dilution, SC-7196, 54 kDa), and HUWE1 rabbit polyclonal antibody (1:500 dilution, SC-134821, 482 kDa). GAPDH rabbit polyclonal antibody (1:10 000 dilution, G9545, 37 kDa) was purchased from Sigma-Aldrich. Proteins were visualized using infrared secondary antibodies (1:10 000) using Li-Cor proprietary software. Relative protein levels were visualized using Li-Cor proprietary software, quantified ImageJ software (National Institutes of Health, Bethesda, MD), and normalized to GAPDH levels within the same lane.
Statistical analysis
For all measurements, data are presented as mean ± standard error of the mean (SEM). All data were analyzed using analysis of variance. Post hoc analysis used the Student-Neuman-Keuls test to detect differences between specific groups. In studies comparing only 2 experimental groups, data were analyzed with a Student t test to determine the significance of treatment effects. Statistical significance was defined as P < .05. Statistical analyses were performed using GraphPad Prism, version 8.0 software (La Jolla, CA).
Results
PPARγ knockdown exacerbates PH and RVH in chimeric SS mouse lungs
We previously reported that Townes SS mice with SCD developed spontaneous PH compared with AA mice.11 Late adult-aged SS mice developed more severe spontaneous PH compared with young-aged SS mice (supplemental Figure 1). To better define the role of endothelial PPARγ function in SCD-PH, we examined novel chimeric mouse models by transplanting bone marrow from SCD mice into mice with endothelial-targeted PPARγ knockout (SSePPARγKO). PH (Figure 1A) and RVH (Figure 1B) were increased in AAePPARγKO or SSLitCon mice compared with AALitCon mice, and PH was exacerbated in SS mice with targeted loss of PPARγ function (SSePPARγKO) compared with SSLitCon mice. As expected, lung levels of PPARγ were reduced in littermate control AAePPARγKO or SSLitCon mice and aggravated in SSePPARγKO mice (Figure 1C) compared with SSLitCon mice. In contrast, compared with AALitCon mice, AAePPARγKO and SSePPARγKO lungs had increased levels of ET-1 (Figure 1D) and VCAM1 (Figure 1E) and that were exacerbated in SSePPARγKO compared with SSLitCon mice. Collectively, these findings indicate that loss of endothelial PPARγ increases expression of markers of endothelial dysfunction and promotes PH in the context of SCD-PH pathogenesis.
![Loss of PPARγ function exacerbates PH and RVH in endothelial-targeted SSePPARγKOchimeric mice. PPARγ levels are decreased, whereas ET-1 and VCAM1 levels are increased in SSePPARγKO mouse lungs. The chimeric SSePPARγKO mice were generated by transplanting SCD bone marrow from Townes mice using previously published methods50-53 into ePPARγKO mice as reported.42,54 ePPARγKO mice were irradiated and transplanted with SS bone marrow. Six weeks after bone marrow transplant, following engraftment, the animals were studied. (A) RVSP was recorded in anesthetized mice with a pressure transducer. Each bar represents the mean RVSP in mm Hg ± SEM; n = 5 to 7. (B) The ratio of the weight of the RV to the LV + septum [RV: (LV + S)] is presented as an index of RVH; n = 5 to 7. (C-E) Whole lung homogenates were collected from littermate control (AALitCon and SSLitCon) and chimeric (AAePPARγKO and SSePPARγKO) mice. Real-time qPCR was performed on lung tissue. Lung PPARγ (C), ET-1 (D), or VCAM (E) levels are expressed relative to 9S normalized to CON values. Each bar represents the mean ± SEM. *P < .05 vs AALitCon; +P < .05 vs SSLitCon, n = 3 to 7.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/2/10.1182_bloodadvances.2020002754/1/m_advancesadv2020002754f1.png?Expires=1767866511&Signature=vAgKhdB03a1ByJQrWrjZPSrMzG8w1v4lloe0l~rl3Xwz5JzXt28PmpIaQRqwGEcZAHAtNOaUO2T-n9VaBXWZLtK8YKiTdT11UcU31j7yQuTrek~ePoRjdmQdCOZRsi9N0h57CP82t46n3l-UWHPXBU0Xf82tBSsBcw1vJdYvQ0xbHlM821epTyModvzhYRexLUDef5oYwvBA9hlGLD8x7eOpng2TXMO1fFQH4zeNo3BD86G9wG9q7AjPbbMsZT06eyWShk-9A3~9G2wvWsPzv~oKRQ7h5t7tPZPJinvsquSzyQTn8yE5AnGlnFWRYvnLfoc1qTz5pU1Darf44zh2WQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Loss of PPARγ function exacerbates PH and RVH in endothelial-targeted SSePPARγKOchimeric mice. PPARγ levels are decreased, whereas ET-1 and VCAM1 levels are increased in SSePPARγKO mouse lungs. The chimeric SSePPARγKO mice were generated by transplanting SCD bone marrow from Townes mice using previously published methods50-53 into ePPARγKO mice as reported.42,54 ePPARγKO mice were irradiated and transplanted with SS bone marrow. Six weeks after bone marrow transplant, following engraftment, the animals were studied. (A) RVSP was recorded in anesthetized mice with a pressure transducer. Each bar represents the mean RVSP in mm Hg ± SEM; n = 5 to 7. (B) The ratio of the weight of the RV to the LV + septum [RV: (LV + S)] is presented as an index of RVH; n = 5 to 7. (C-E) Whole lung homogenates were collected from littermate control (AALitCon and SSLitCon) and chimeric (AAePPARγKO and SSePPARγKO) mice. Real-time qPCR was performed on lung tissue. Lung PPARγ (C), ET-1 (D), or VCAM (E) levels are expressed relative to 9S normalized to CON values. Each bar represents the mean ± SEM. *P < .05 vs AALitCon; +P < .05 vs SSLitCon, n = 3 to 7.
Loss of PPARγ function exacerbates PH and RVH in endothelial-targeted SSePPARγKOchimeric mice. PPARγ levels are decreased, whereas ET-1 and VCAM1 levels are increased in SSePPARγKO mouse lungs. The chimeric SSePPARγKO mice were generated by transplanting SCD bone marrow from Townes mice using previously published methods50-53 into ePPARγKO mice as reported.42,54 ePPARγKO mice were irradiated and transplanted with SS bone marrow. Six weeks after bone marrow transplant, following engraftment, the animals were studied. (A) RVSP was recorded in anesthetized mice with a pressure transducer. Each bar represents the mean RVSP in mm Hg ± SEM; n = 5 to 7. (B) The ratio of the weight of the RV to the LV + septum [RV: (LV + S)] is presented as an index of RVH; n = 5 to 7. (C-E) Whole lung homogenates were collected from littermate control (AALitCon and SSLitCon) and chimeric (AAePPARγKO and SSePPARγKO) mice. Real-time qPCR was performed on lung tissue. Lung PPARγ (C), ET-1 (D), or VCAM (E) levels are expressed relative to 9S normalized to CON values. Each bar represents the mean ± SEM. *P < .05 vs AALitCon; +P < .05 vs SSLitCon, n = 3 to 7.
HUWE1 and miR-98 are reduced in the lungs of SCD mice and in HEM-treated HPAECs
We have previously demonstrated that reductions in PPARγ increase ET-1 by decreasing miR-98 levels in hypoxia-induced PH models.42 Because in silico analysis indicates that HUWE1 serves as the host gene for miR-98 (Figure 2A), we examined the levels of HUWE1 and miR-98 in SS mouse lungs and HEM-treated HPAECs. Levels of HUWE1 mRNA and protein (Figure 2B) and miR-98 (Figure 2C) were significantly decreased in the lungs of SS compared with AA mice. Similarly, treatment with HEM (5-10 µM) for 72 hours reduced HPAEC HUWE1 mRNA and protein (Figure 2D) and miR-98 (Figure 2E) levels in vitro. Furthermore, treatment with 5 µM HEM significantly reduced HUWE1 mRNA and protein (Figure 2F) and miR-98 (Figure 2G) levels after 48 or 72 hours, but not after 24 hours.
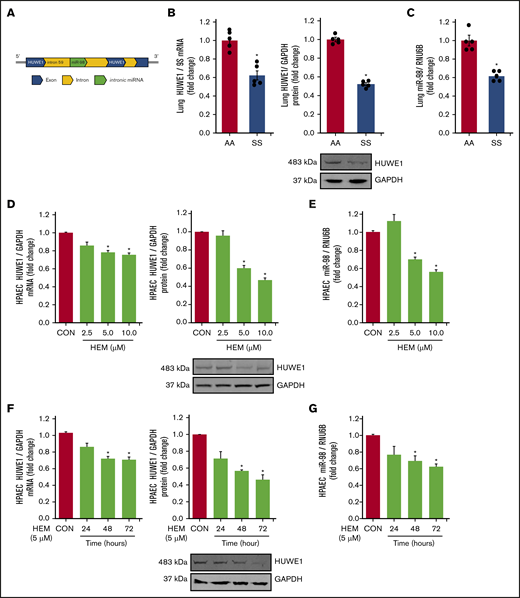
HUWE1 and miR-98 levels are reduced in lungs of SS mice in vivo and in HEM-treated HPAECs in vitro. (A) Schematic illustration of intronic miR-98 on the host gene HUWE1 on chromosome X. (B-C) Whole lung homogenates were collected from littermate control (AA) and SS mice. Lung HUWE1 mRNA and protein (B) and miR-98 (C) levels were measured with real-time qPCR or western blotting and expressed relative to lung mRNA (9S mRNA), protein (GAPDH), and RNU6B (miRNA). *P < .05 vs AA, n = 5. (D-E) HPAECs were treated with DMSO vehicle (CON) or HEM (2.5, 5.0, and 10.0 µM) for 72 hours. Mean HPAEC HUWE1 mRNA and protein (D) and miR-98 (E) levels were measured with real-time qPCR or western blotting. (F-G) HPAECs were treated with DMSO vehicle (CON) or HEM (5 µM) for 24, 48, and 72 hours. Mean HPAEC HUWE1 mRNA and protein (F) and miR-98 (G) levels were measured with real-time qPCR or western blotting. Each bar represents the mean HUWE1 and miR-98 level ± SEM relative to GAPDH or RNU6B expressed as fold change vs CON. *P < .05 vs CON, n = 6.
HUWE1 and miR-98 levels are reduced in lungs of SS mice in vivo and in HEM-treated HPAECs in vitro. (A) Schematic illustration of intronic miR-98 on the host gene HUWE1 on chromosome X. (B-C) Whole lung homogenates were collected from littermate control (AA) and SS mice. Lung HUWE1 mRNA and protein (B) and miR-98 (C) levels were measured with real-time qPCR or western blotting and expressed relative to lung mRNA (9S mRNA), protein (GAPDH), and RNU6B (miRNA). *P < .05 vs AA, n = 5. (D-E) HPAECs were treated with DMSO vehicle (CON) or HEM (2.5, 5.0, and 10.0 µM) for 72 hours. Mean HPAEC HUWE1 mRNA and protein (D) and miR-98 (E) levels were measured with real-time qPCR or western blotting. (F-G) HPAECs were treated with DMSO vehicle (CON) or HEM (5 µM) for 24, 48, and 72 hours. Mean HPAEC HUWE1 mRNA and protein (F) and miR-98 (G) levels were measured with real-time qPCR or western blotting. Each bar represents the mean HUWE1 and miR-98 level ± SEM relative to GAPDH or RNU6B expressed as fold change vs CON. *P < .05 vs CON, n = 6.
NF-κB/p65, ET-1, and endothelial dysfunction are increased in lungs of SS mice in vivo and in HEM-treated HPAECs in vitro
Among the derangements caused by intravascular hemolysis in SCD, the release of free heme increases NF-κB/p65 activation and induces endothelial dysfunction.59,60 Loss of PPARγ function also increases p65,45 which stimulates ET-142 and adhesion molecule expression. These observations suggest that both increases in HEM and reductions in PPARγ may regulate the expression of NF-κB/p65 in SCD. Levels of p65 (Figure 3A) and VCAM1 (supplemental Figure 2A) mRNA and protein were increased in the lungs of SS compared with AA mice. In vitro, HEM (5.0-10.0 µM) stimulated p65 (Figure 3B), ET-1 (Figure 3C), and VCAM1 expression (supplemental Figure 2B) and HPAEC monocyte adhesion and proliferation (supplemental Figure 2C-D) indicative of endothelial dysfunction. Furthermore, treatment with HEM for 48 to 72 but not 24 hours increased HPAEC p65 mRNA and protein (Figure 3D) and ET-1 mRNA (Figure 3E).
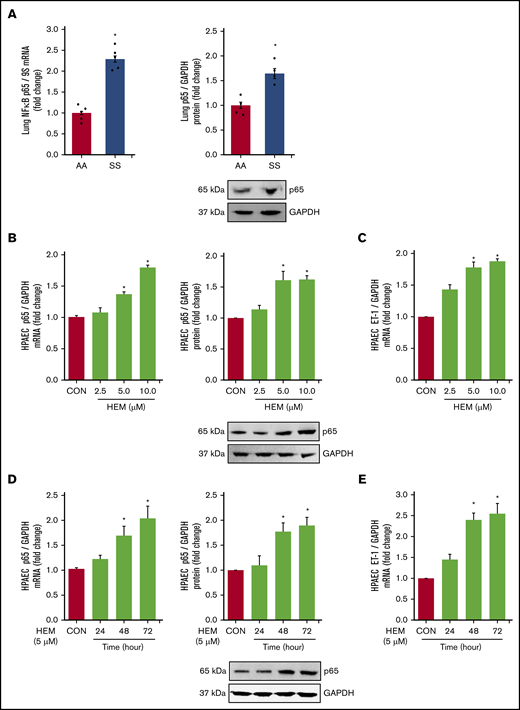
NF-κB/p65 levels are increased and HUWE1 levels are reduced in lungs of SS mice in vivo and in HEM-treated HPAECs in vitro. (A) Whole lung homogenates were collected from littermate control (AA) and SS mice. Lung NF-κB/p65 mRNA or protein levels were measured with real-time qPCR or western blotting and expressed relative to lung mRNA (9S mRNA) or protein (GAPDH). *P < .05 vs AA, n = 4 to 5. (B-C) HPAECs were treated with DMSO vehicle (CON) or HEM (2.5, 5.0, and 10.0 µM) for 72 hours. (D-E) HPAECs were treated with DMSO vehicle (CON) or HEM (5 µM) for 24, 48, and 72 hours. Mean HPAEC NF-κB/p65 mRNA or protein (B) and ET-1 mRNA (C) levels were measured with real-time qPCR or western blotting. Each bar represents the mean NF-κB/p65 or ET-1 level ± SEM relative to GAPDH expressed as fold change vs CON. *P < .05 vs CON, n = 6. (D-E) HPAECs were treated with DMSO vehicle (CON) or HEM (5 µM) for 24, 48, and 72 hours. Mean HPAEC p65 mRNA and protein (D) and ET-1 (E) protein levels were measured with real-time qPCR or western blotting. Each bar represents the mean p65 or ET-1 level ± SEM relative to GAPDH expressed as fold change vs CON. *P < .05 vs CON, n = 6.
NF-κB/p65 levels are increased and HUWE1 levels are reduced in lungs of SS mice in vivo and in HEM-treated HPAECs in vitro. (A) Whole lung homogenates were collected from littermate control (AA) and SS mice. Lung NF-κB/p65 mRNA or protein levels were measured with real-time qPCR or western blotting and expressed relative to lung mRNA (9S mRNA) or protein (GAPDH). *P < .05 vs AA, n = 4 to 5. (B-C) HPAECs were treated with DMSO vehicle (CON) or HEM (2.5, 5.0, and 10.0 µM) for 72 hours. (D-E) HPAECs were treated with DMSO vehicle (CON) or HEM (5 µM) for 24, 48, and 72 hours. Mean HPAEC NF-κB/p65 mRNA or protein (B) and ET-1 mRNA (C) levels were measured with real-time qPCR or western blotting. Each bar represents the mean NF-κB/p65 or ET-1 level ± SEM relative to GAPDH expressed as fold change vs CON. *P < .05 vs CON, n = 6. (D-E) HPAECs were treated with DMSO vehicle (CON) or HEM (5 µM) for 24, 48, and 72 hours. Mean HPAEC p65 mRNA and protein (D) and ET-1 (E) protein levels were measured with real-time qPCR or western blotting. Each bar represents the mean p65 or ET-1 level ± SEM relative to GAPDH expressed as fold change vs CON. *P < .05 vs CON, n = 6.
HUWE1 induces NF-κB/p65 degradation
To further dissect the relationship between p65 and HUWE1, we screened 3 small interfering RNAs (siRNAs; 10 nM) to HUWE1 (NM_031407) to knockdown HUWE1 (supplemental Figure 3) to levels comparable to those in HEM-treated HPAEC. We selected siHUWE1-V2, which targeted the exon 36 to 37 region of the HUWE1 protein coding sequence (Figure 4A). As illustrated in Figure 4B, reductions in HUWE1 are sufficient to increase HPAEC p65 protein levels. These findings support novel SCD-related pathways that contribute to increased p65 signaling and endothelial dysfunction. To further explore if HUWE1 mediates p65 proteasomal degradation, p65 was immunoprecipitated from HPAECs treated with scrambled siRNA or siHUWE1 plus CHX to inhibit protein synthesis. As shown in Figure 4C, depletion of HUWE1 attenuated decreases in p65 protein half-life. These findings suggest that HUWE1 decreased the half-life of p65 protein, consistent with HUWE1-mediated p65 ubiquitination and degradation. Importantly, overexpression of HUWE1 attenuated HEM-induced p65, ET-1, and VCAM1 levels in HPAECs (Figure 4D-G), suggesting that HUWE1-induced p65 degradation was involved in p65 proteasome-dependent degradation.
![HUWE1, E3 ligase induces the degradation of NF-κB/p65. (A-B) HPAECs were treated with scrambled (SCR) or HUWE1 (10 or 20 nM) siRNAs for 6 hours and then incubated for an additional 72 hours. Real-time qPCR or western blotting was performed for HUWE1 mRNA (A) or p65 protein (B). Each bar represents mean ± SEM HUWE1 or p65 level relative to GAPDH expressed as fold change vs cells treated with scrambled siRNA (SCR). *P < .05 vs SCR, n = 4 to 6. (C) HPAECs were transfected with scrambled siRNA or HUWE1 siRNA for 72 hours and were treated with CHX (40 µg/mL) for 0, 2, 4, and 8 hours to inhibit de novo protein synthesis and harvested for western blotting. The levels of p65 at time 0 was set as 100% and the percent p65 protein remaining following CHX treatment at each time point was calculated accordingly. (D-F) HPAECs were transfected with either the CON plasmid (VEC or HEM/oxHUWE1[−]) or HUWE1 (1 μg, oxHUWE1 or HEM/oxHUWE1[+]) plasmid for 6 hours and then treated with DMSO vehicle (CON) or HEM (5 µM) for 72 hours. Mean HPAEC HUWE1 (D), p65 (E), ET-1 (F), and VCAM1 (G) mRNA levels were measured with real-time qPCR or western blotting. Each bar represents the mean mRNA level ± SEM relative to GAPDH expressed as fold change vs CON. *P < .05 vs VEC or HEM/oxHUWE1−, n = 6.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/2/10.1182_bloodadvances.2020002754/1/m_advancesadv2020002754f4.png?Expires=1767866511&Signature=ScVTxbEcC7bGgr2EwK8LBkeWR3QedvxMl1f~I5a5votpsNVwG59sTpczfgmtD3~jCUkBbD485qFAb61qcobwQ99Fyx1XtD9Kxyikad3fySfAGO7vtXfZM1x1MvcO1aXSLmU9LRP-ht7EqGgbm5LgOXrwGwA6kM3zPn97TVLUDivRfuDpBOzJImCo2bN2u62WGEEscAbuSBVLxNKJTRVUd~dHvUicM-SAfUySL8AG8Bx9ossCuRVzZqRD71RYp3Tq9JT7rWnCD5-N-lVJ5c8fK9MuV7zkN1Jr3CyvPLl5La6KAD3AZRcFnq0rF7txhwt9u7j8mLwTDr7aTFxSQhJYvw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
HUWE1, E3 ligase induces the degradation of NF-κB/p65. (A-B) HPAECs were treated with scrambled (SCR) or HUWE1 (10 or 20 nM) siRNAs for 6 hours and then incubated for an additional 72 hours. Real-time qPCR or western blotting was performed for HUWE1 mRNA (A) or p65 protein (B). Each bar represents mean ± SEM HUWE1 or p65 level relative to GAPDH expressed as fold change vs cells treated with scrambled siRNA (SCR). *P < .05 vs SCR, n = 4 to 6. (C) HPAECs were transfected with scrambled siRNA or HUWE1 siRNA for 72 hours and were treated with CHX (40 µg/mL) for 0, 2, 4, and 8 hours to inhibit de novo protein synthesis and harvested for western blotting. The levels of p65 at time 0 was set as 100% and the percent p65 protein remaining following CHX treatment at each time point was calculated accordingly. (D-F) HPAECs were transfected with either the CON plasmid (VEC or HEM/oxHUWE1[−]) or HUWE1 (1 μg, oxHUWE1 or HEM/oxHUWE1[+]) plasmid for 6 hours and then treated with DMSO vehicle (CON) or HEM (5 µM) for 72 hours. Mean HPAEC HUWE1 (D), p65 (E), ET-1 (F), and VCAM1 (G) mRNA levels were measured with real-time qPCR or western blotting. Each bar represents the mean mRNA level ± SEM relative to GAPDH expressed as fold change vs CON. *P < .05 vs VEC or HEM/oxHUWE1−, n = 6.
HUWE1, E3 ligase induces the degradation of NF-κB/p65. (A-B) HPAECs were treated with scrambled (SCR) or HUWE1 (10 or 20 nM) siRNAs for 6 hours and then incubated for an additional 72 hours. Real-time qPCR or western blotting was performed for HUWE1 mRNA (A) or p65 protein (B). Each bar represents mean ± SEM HUWE1 or p65 level relative to GAPDH expressed as fold change vs cells treated with scrambled siRNA (SCR). *P < .05 vs SCR, n = 4 to 6. (C) HPAECs were transfected with scrambled siRNA or HUWE1 siRNA for 72 hours and were treated with CHX (40 µg/mL) for 0, 2, 4, and 8 hours to inhibit de novo protein synthesis and harvested for western blotting. The levels of p65 at time 0 was set as 100% and the percent p65 protein remaining following CHX treatment at each time point was calculated accordingly. (D-F) HPAECs were transfected with either the CON plasmid (VEC or HEM/oxHUWE1[−]) or HUWE1 (1 μg, oxHUWE1 or HEM/oxHUWE1[+]) plasmid for 6 hours and then treated with DMSO vehicle (CON) or HEM (5 µM) for 72 hours. Mean HPAEC HUWE1 (D), p65 (E), ET-1 (F), and VCAM1 (G) mRNA levels were measured with real-time qPCR or western blotting. Each bar represents the mean mRNA level ± SEM relative to GAPDH expressed as fold change vs CON. *P < .05 vs VEC or HEM/oxHUWE1−, n = 6.
NF-κB/p65 is degraded by ubiquitination
To determine whether p65 is degraded by ubiquitination, HPAECs were transfected with either CON plasmids (empty VEC) or 1 µg of HA-ubiquitin plasmids (HA-tagged Ub). Ectopically expressed HA-tagged Ub plasmid increased HPAEC ubiquitin levels and decreased endogenous p65 levels (Figure 5A). To further examine if HUWE1 acts as an E3 ligase to mediate p65 ubiquitination, HPAECs were transfected with either CON plasmids (empty VEC) or 1 µg of HUWE1 plasmids. Overexpression of HUWE1 directly induced p65 degradation (Figure 5B-C), indicating that HUWE1 ubiquitinates p65. To further dissect mechanisms of p65 protein degradation, HPAECs were treated with graded concentrations and durations of CHX. As illustrated in Figure 5D, CHX (80 μg/mL) treatment sharply reduced the half-life of p65. To determine whether p65 degradation is primarily dependent on the ubiquitin proteasome system or endosome-lysosome pathway, HPAECs were pretreated with the proteasome inhibitor, MG132, or with the lysosome inhibitor, leupeptin, for 2 hours after CHX treatment. MG132, but not leupeptin, stabilized p65 protein levels, consistent with ubiquitin proteasome, rather than lysosomal-mediated p65 degradation in HPAECs (Figure 5E). These results suggest that reductions in HUWE1 in SS lung or HEM-treated HPAECs could contribute to reduced p65 ubiquitination and degradation resulting in upregulation of p65 and its downstream targets. As illustrated by p65 knockdown studies, p65 stimulates the expression of mediators of endothelial dysfunction, including ET-1 and VCAM1 levels (supplemental Figure 4A-C), whereas silencing of p65 had no effect on HUWE1 levels (data not shown).
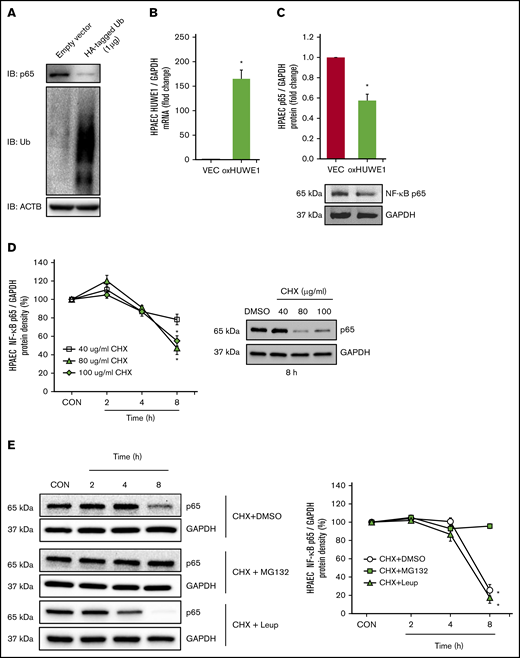
NF-κB/p65 is degraded by ubiquitination. (A) HPAECs were transfected with either the CON plasmid (empty VEC) or HA-ubiquitin (HA-Ubi; 1 µg) plasmid for 24 hours. HPAECs were collected and assayed for HA, p65, and β-actin (loading CON, ACTB) by immunoblotting. (B-C) HPAECs were transfected with either the CON plasmid (VEC) or HUWE1 (1 μg, oxHUWE1) plasmid for 6 hours; Media were replaced with EGM containing 5% FBS and then incubated for an additional 72 hours. (D) CHX treatment was carried out at a concentration of 40 to 100 μg/mL at varying time points (0-8 hours) in 0% FBS medium. (E) HPAECs were pretreated with DMSO or MG132, and leupeptin (Leup) for 2 hours and treated with CHX (40 µg/mL) for 0, 2, 4, and 8 hours to inhibit de novo protein synthesis and harvested for western blotting. The levels of p65 at time 0 was set as 100%, and the percent p65 protein remaining following CHX treatment at each time point was calculated accordingly. ACTB, actin beta.
NF-κB/p65 is degraded by ubiquitination. (A) HPAECs were transfected with either the CON plasmid (empty VEC) or HA-ubiquitin (HA-Ubi; 1 µg) plasmid for 24 hours. HPAECs were collected and assayed for HA, p65, and β-actin (loading CON, ACTB) by immunoblotting. (B-C) HPAECs were transfected with either the CON plasmid (VEC) or HUWE1 (1 μg, oxHUWE1) plasmid for 6 hours; Media were replaced with EGM containing 5% FBS and then incubated for an additional 72 hours. (D) CHX treatment was carried out at a concentration of 40 to 100 μg/mL at varying time points (0-8 hours) in 0% FBS medium. (E) HPAECs were pretreated with DMSO or MG132, and leupeptin (Leup) for 2 hours and treated with CHX (40 µg/mL) for 0, 2, 4, and 8 hours to inhibit de novo protein synthesis and harvested for western blotting. The levels of p65 at time 0 was set as 100%, and the percent p65 protein remaining following CHX treatment at each time point was calculated accordingly. ACTB, actin beta.
PPARγ regulates HUWE1 expression
In silico analysis revealed putative PPAR response elements within the HUWE1 promoter region. We previously reported that adenoviral-mediated PPARγ overexpression increased miR-98 and decreased ET-1 levels in HPAECs in vitro and that endothelial-targeted PPARγ overexpression (ePPARγOX) in mice increased miR-98 levels in lung homogenates in vivo.42 Consistent with those findings, PPARγ overexpression upregulated HUWE1 levels in vitro (Figure 6A) and in vivo (Figure 6B), attenuated HEM-induced reductions in HPAEC HUWE1 (Figure 6C) and miR-98 (Figure 6D) levels, and attenuated basal as well as HEM-induced increases in p65 levels (Figure 6E). In contrast, compared with littermate control AA (AALitCon) mice, levels of HUWE1 (Figure 6F) and miR-9842 (Figure 6G) were reduced in lung homogenates from endothelial-targeted PPARγ KO (AAePPARγKO) and SSLitCon mice in vivo. Similarly, ∼60% depletion of PPARγ in HPAECs in vitro (supplemental Figure 5A) attenuated HPAEC HUWE1 and miR-98 levels (supplemental Figure 5B-C). Furthermore, lung levels of HUWE1 (Figure 6F) and miR-98 (Figure 6G) were reduced in SSePPARγKO mice compared with SSLitCon mice, whereas SSePPARγKO lungs were characterized by increased levels of p65 (Figure 6H). Collectively, these findings indicate that loss of endothelial PPARγ is sufficient to reduce HUWE1 and miR-98, leading to increases in p65 in SCD-PH pathogenesis.
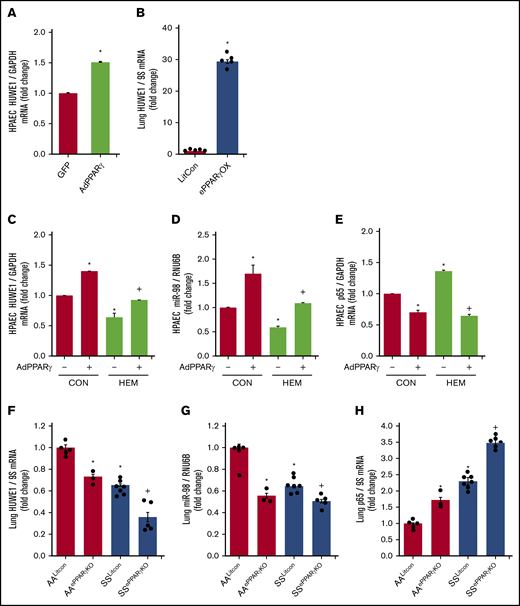
PPARγ activates HUWE1 expression and decreases p65 levels. (A,C-E) HPAECs were transfected with green fluorescent protein (GFP) or AdPPARγ (25 multiplicity of infection) constructs for PPARγ overexpression. After 6 hours, HPAECs were then incubated for an additional 72 hours with DMSO vehicle (CON) or HEM (5 µM). Real-time qPCR was performed for HUWE1 (A,C), miR-98 (D), or p65 (E). Each bar represents mean ± SEM HUWE1, miR-98, or p65 level relative to GAPDH or RNU6B expressed as fold change vs cells treated with GFP. *P < .05 vs GFP or CON/AdPPARγ(−); +P < .05 vs HEM/AdPPARγ(−), n = 5 to 6. (B) Whole lungs were collected from littermate control (FulCon) or endothelial-targeted PPARγ overexpression (ePPARγOX) mice. Levels of lung HUWE1 were measured with real-time qPCR and expressed relative to lung GPADH mRNA. *P < .05 vs FulCon, n = 6. (F-H) The chimeric SSePPARγKO mice were generated by transplanting SCD bone marrow from Townes mice using methods we previously published50-53 into ePPARγKO mice as we have reported.42,54 ePPARγKO mice were irradiated and transplanted with SS bone marrow. Whole lung homogenates were collected from littermate control (AALitCon and SSLitCon) and chimeric (AAePPARγKO and SSePPARγKO) mice. Real-time qPCR was performed on lung tissue. Lung HUWE1 (F), miR-98 (G), or p65 (H) levels are expressed relative to 9S or RNU6B normalized to CON values. Each bar represents the mean ± SEM. *P < .05 vs AALitCon; +P < .05 vs + SSLitCon, n = 3 to 7.
PPARγ activates HUWE1 expression and decreases p65 levels. (A,C-E) HPAECs were transfected with green fluorescent protein (GFP) or AdPPARγ (25 multiplicity of infection) constructs for PPARγ overexpression. After 6 hours, HPAECs were then incubated for an additional 72 hours with DMSO vehicle (CON) or HEM (5 µM). Real-time qPCR was performed for HUWE1 (A,C), miR-98 (D), or p65 (E). Each bar represents mean ± SEM HUWE1, miR-98, or p65 level relative to GAPDH or RNU6B expressed as fold change vs cells treated with GFP. *P < .05 vs GFP or CON/AdPPARγ(−); +P < .05 vs HEM/AdPPARγ(−), n = 5 to 6. (B) Whole lungs were collected from littermate control (FulCon) or endothelial-targeted PPARγ overexpression (ePPARγOX) mice. Levels of lung HUWE1 were measured with real-time qPCR and expressed relative to lung GPADH mRNA. *P < .05 vs FulCon, n = 6. (F-H) The chimeric SSePPARγKO mice were generated by transplanting SCD bone marrow from Townes mice using methods we previously published50-53 into ePPARγKO mice as we have reported.42,54 ePPARγKO mice were irradiated and transplanted with SS bone marrow. Whole lung homogenates were collected from littermate control (AALitCon and SSLitCon) and chimeric (AAePPARγKO and SSePPARγKO) mice. Real-time qPCR was performed on lung tissue. Lung HUWE1 (F), miR-98 (G), or p65 (H) levels are expressed relative to 9S or RNU6B normalized to CON values. Each bar represents the mean ± SEM. *P < .05 vs AALitCon; +P < .05 vs + SSLitCon, n = 3 to 7.
PPARγ activation attenuates both reductions in HUWE1 and miR-98 in vitro and increases in PH and RVH in vivo
We previously demonstrated that Townes SS mice develop PH and RVH.11 We also reported that pharmacological PPARγ activation attenuates (1) reductions in miR-98 levels,42 and (2) increases in ET-1 and NF-κB signaling in hypoxia-induced PH in mice.46 We now extend these observations to the pathobiology of SCD-PH by demonstrating that PPARγ activation using RSG (1) attenuated baseline PH (Figure 7A) and RVH (Figure 7B) in SS mice, (2) increased HUWE1 (Figure 7C) and miR-98 (Figure 7D) levels in SS mouse lungs, and (3) attenuated p65 (Figure 7E) and ET-1 (Figure 7F) levels. Similarly, in HEM-treated HPAECs in vitro, PPARγ activation restored levels of HUWE1 (Figure 7G) and miR-98 (Figure 7H) and attenuated increases in NF-κB/p65 (Figure 7I), ET-1 (Figure 7J), and VCAM1 (supplemental Figure 6A). To examine more functional endothelial endpoints, the effect of RSG on HEM-induced alterations in HPAEC monocyte adhesion and cell proliferation was determined. HEM-stimulated monocyte adhesion (supplemental Figure 6B) and HPAEC proliferation (supplemental Figure 6C) were attenuated by RSG treatment. Furthermore, overexpression of miR-98 using miR-98 mimic attenuated increases in ET-1 levels (supplemental Figure 6D). Collectively, these findings indicate that SCD-induced hemolysis decreases the PPARγ/HUWE1/miR-98 axis to increase p65 and that intervention with PPARγ activation attenuates these derangements.
![The PPARγ ligand, RSG, attenuates decreases in HUWE1 and miR-98 levels and increases in p65 and ET-1 levels in SS mouse lung and in HEM-treated HPAECs. (A) RVSP was recorded in anesthetized mice with a pressure transducer. Each bar represents the mean RVSP in mm Hg ± SEM; n = 5 to 6. (B) The ratio of the weight of the RV to the LV + septum [RV: (LV + S)] is presented as an index of RVH. n = 5 to 6. (C-F) Whole lung homogenates were collected from (AA) and SS mice following gavage with RSG (10 mg/kg per day) or vehicle for 10 days. Real-time qPCR was performed on lung tissue. Lung HUWE1 (C), miR-98 (D), p65 (E), or ET-1 (F) levels are expressed relative to GAPDH or RNU6B and normalized to CON values. Each bar represents the mean ± SEM. *P < .05 vs AA; +P < .05 vs SS, n = 5 to 6. (G-J) HPAECs were treated with HEM (5 µM) for 72 hours. During the final 24 hours of HEM exposure, selected HPAECs were treated ± RSG (10 μM). Real-time qPCR was performed for HUWE1 (G), miR-98 (H), p65 (I), or ET-1 (J) levels. Each bar represents the mean ± SEM relative to RNU6B or GAPDH as indicated. *P < .05 vs HEM/RSG(−), n = 3 to 6. (K) Hypothetical schema defining the role of PPARγ/HUWE1/miR-98 signaling in SCD-PH pathogenesis. Hemolysis induces reductions in PPARγ that decrease miR-98 and HUWE1 levels. Reductions in miR-98 stimulate ET-1 and reductions in HUWE1 increase NF-κB, adhesion molecule expression, and endothelial dysfunction promoting SCD-PH pathogenesis.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/2/10.1182_bloodadvances.2020002754/1/m_advancesadv2020002754f7.png?Expires=1767866511&Signature=F-604fblAzyNaUUDXLmFk-Pan96FYxBORQcwULO2aJQKIKtMqr9SpG7GN3wpEBPjgW3f1dAri7mfwp~2htaZc0-iGNG-N3qoua0YkBfFGHyeC7-C77kbb0sjAlK9jVDjtU2qTMIaorFmyvUUaOfbzd52j46N1lytguEzco1N8cVezg0ZZygrwpb1T6aAvnzO36Edduqw8kth5kACbq9nhbdTKCtEb0YQ38t2Vpvq51hWoMvUXnNpuuL0bR~TXNPHrfGCLo~7TTx5HqCmi9DAZ4uNWNBjTjxvSUE3Ka2N2uug1Jf46u-e9UJiJGPD1CwrrNY2O8BExpjBDiAxAHJpAA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The PPARγ ligand, RSG, attenuates decreases in HUWE1 and miR-98 levels and increases in p65 and ET-1 levels in SS mouse lung and in HEM-treated HPAECs. (A) RVSP was recorded in anesthetized mice with a pressure transducer. Each bar represents the mean RVSP in mm Hg ± SEM; n = 5 to 6. (B) The ratio of the weight of the RV to the LV + septum [RV: (LV + S)] is presented as an index of RVH. n = 5 to 6. (C-F) Whole lung homogenates were collected from (AA) and SS mice following gavage with RSG (10 mg/kg per day) or vehicle for 10 days. Real-time qPCR was performed on lung tissue. Lung HUWE1 (C), miR-98 (D), p65 (E), or ET-1 (F) levels are expressed relative to GAPDH or RNU6B and normalized to CON values. Each bar represents the mean ± SEM. *P < .05 vs AA; +P < .05 vs SS, n = 5 to 6. (G-J) HPAECs were treated with HEM (5 µM) for 72 hours. During the final 24 hours of HEM exposure, selected HPAECs were treated ± RSG (10 μM). Real-time qPCR was performed for HUWE1 (G), miR-98 (H), p65 (I), or ET-1 (J) levels. Each bar represents the mean ± SEM relative to RNU6B or GAPDH as indicated. *P < .05 vs HEM/RSG(−), n = 3 to 6. (K) Hypothetical schema defining the role of PPARγ/HUWE1/miR-98 signaling in SCD-PH pathogenesis. Hemolysis induces reductions in PPARγ that decrease miR-98 and HUWE1 levels. Reductions in miR-98 stimulate ET-1 and reductions in HUWE1 increase NF-κB, adhesion molecule expression, and endothelial dysfunction promoting SCD-PH pathogenesis.
The PPARγ ligand, RSG, attenuates decreases in HUWE1 and miR-98 levels and increases in p65 and ET-1 levels in SS mouse lung and in HEM-treated HPAECs. (A) RVSP was recorded in anesthetized mice with a pressure transducer. Each bar represents the mean RVSP in mm Hg ± SEM; n = 5 to 6. (B) The ratio of the weight of the RV to the LV + septum [RV: (LV + S)] is presented as an index of RVH. n = 5 to 6. (C-F) Whole lung homogenates were collected from (AA) and SS mice following gavage with RSG (10 mg/kg per day) or vehicle for 10 days. Real-time qPCR was performed on lung tissue. Lung HUWE1 (C), miR-98 (D), p65 (E), or ET-1 (F) levels are expressed relative to GAPDH or RNU6B and normalized to CON values. Each bar represents the mean ± SEM. *P < .05 vs AA; +P < .05 vs SS, n = 5 to 6. (G-J) HPAECs were treated with HEM (5 µM) for 72 hours. During the final 24 hours of HEM exposure, selected HPAECs were treated ± RSG (10 μM). Real-time qPCR was performed for HUWE1 (G), miR-98 (H), p65 (I), or ET-1 (J) levels. Each bar represents the mean ± SEM relative to RNU6B or GAPDH as indicated. *P < .05 vs HEM/RSG(−), n = 3 to 6. (K) Hypothetical schema defining the role of PPARγ/HUWE1/miR-98 signaling in SCD-PH pathogenesis. Hemolysis induces reductions in PPARγ that decrease miR-98 and HUWE1 levels. Reductions in miR-98 stimulate ET-1 and reductions in HUWE1 increase NF-κB, adhesion molecule expression, and endothelial dysfunction promoting SCD-PH pathogenesis.
Discussion
In the current study, we demonstrate for the first time that (1) reductions in PPARγ contribute to pulmonary vascular alterations in SCD-PH pathogenesis, (2) HUWE1 and miR-98 levels are reduced in lungs from SS mice and in HEM-treated HPAECs, (3) HUWE1, an E3 ligase, induces ubiquitination and proteasomal degradation of p65, (4) PPARγ activation increases HUWE1 and miR-98 levels, and (5) targeting PPARγ activation provides a previously unrecognized therapeutic strategy for SCD-PH that merits additional investigation. Although the pathobiology of SCD-PH is undoubtedly complicated, involving multiple pathways, our results provide evidence that reductions in PPARγ increase levels of p65 and downstream targets, ET-1 and VCAM1, that promote endothelial dysfunction in vivo and in vitro. In concordance with these findings, PH, RVH, p65, and ET-1 levels are increased in novel chimeric SS mice with endothelial-targeted PPARγ loss of function (SSePPARγKO), where levels of HUWE1 and miR-98 are reduced. However, activation of PPARγ with RSG or adenoviral-mediated PPARγ overexpression attenuates these derangements in part through reductions in NF-κB and ET-1, suggesting that therapeutically targeting PPARγ-HUWE1-miR-98 signaling provides a novel strategy for reducing SCD-associated pulmonary endothelial dysfunction and PH (Figure 7K).
To rigorously investigate the role of PPARγ in SCD-PH, we examined novel chimeric mouse models generated by transplanting bone marrow from Townes SCD mice into endothelial-targeted PPARγ gain (SSePPARγOX) or loss (SSePPARγKO) of function models.61 HEM or SCD-associated alterations in endothelial gene expression were attenuated in models with enhanced PPARγ function and aggravated under conditions of reduced PPARγ function. These findings further define novel molecular and cellular mechanisms for endothelial dysfunction in SCD-PH and provide additional rationale for exploring PPARγ as a novel therapeutic target in SCD-PH. Our findings also illustrate that aging in this transgenic SCD model appears to be associated with worsening PH phenotype (supplemental Figure 1), a feature that mirrors clinical observations of enhanced PH risk in aging SCD patients.55 Although our in vitro studies indicate that HEM recapitulates many of the molecular signatures of in vivo SCD-PH, other products of intravascular hemolysis55,59,60 undoubtedly contribute to these derangements.
As an E3 ubiquitin ligase, HUWE1 plays an important role in apoptosis, DNA replication, and DNA damage repair targeting proteins for proteasomal degradation. In silico analysis of the HUWE1 promoter revealed putative PPARγ but not NF-κB binding sites. We previously demonstrated that PPARγ loss of function increased NF-κB/p65 in hypoxia-induced PH, whereas PPARγ gain of function attenuated NF-κB levels.46 In addition, we and others demonstrated that PPARγ negatively regulates p65 expression through transrepression mechanisms.45,47 PPARγ can also induce ubiquitination and degradation of p65.48 The current study demonstrates that PPARγ activation increased HUWE1 levels (Figure 6). These findings led us to postulate that chronic hemolysis in SCD stimulates loss of PPARγ that causes reductions in HUWE1 and its E3 ligase activity, leading to reduced ubiquitination and degradation of p65. The current study uncovers a novel mechanism whereby HUWE1, acting as an E3 ligase, induces ubiquitination of p65 leading to inhibition of NF-κB signaling and reductions in ET-1, endothelial dysfunction, and SCD-PH pathogenesis (Figures 4 and 5).
PPARγ is reduced in the lung or pulmonary vascular tissues of virtually every model of PH in which it has been examined.26 Several mechanisms have been described that contribute to reductions in PPARγ in PH. For example, we reported increased lung levels of miR-27a in Townes mice with SCD-PH.11 miR-27a binds the 3′ untranslated region of PPARγ to reduce PPARγ levels in the lung.54 In contrast, PPARγ activation with RSG attenuated reductions in PPARγ and increases in miR-27a, ET-1, and markers of endothelial dysfunction. However, the ability of PPARγ activation to attenuate SCD-PH has not been previously established, and underlying therapeutic mechanisms had not been defined. The current study provides novel evidence that miR-98 is reduced in the lungs of SS compared with AA mice (Figure 2). Furthermore, PPARγ activation attenuated reductions in miR-98 levels, PH, and RVH. The current study also demonstrates that activating PPARγ prevents decreases in miR-98 and attenuates HEM-induced increases in ET-1. These findings suggest a novel paradigm in which activation of PPARγ favorably modulates programs of gene expression in SCD-PH that contribute to endothelial dysfunction.
Significant alterations in ubiquitin-proteasome–mediated protein degradation have been reported to participate in PH pathogenesis.62 The current findings link reductions in PPARγ to reduced HUWE1 expression through reduced PPARγ-stimulated HUWE1 activation. Our data indicate that loss of HUWE1 E3 ligase activity increases p65 transcription and decreases its E3 ligase-mediated proteasomal degradation of p65 leading to (1) increased ET-1 expression, an important stimulus for vasoconstriction, pulmonary vascular cell proliferation, and therapeutic target in PH,46 and (2) increased VCAM1 expression, which promotes endothelial monocyte adhesion and lung inflammation.
In summary, to our knowledge, the current study provides the first evidence that chronic hemolysis reduces HUWE1 and miR-98 by suppression of PPARγ expression and increases NF-κB/p65 through posttranslational mechanisms in vivo and in vitro, leading to increases in ET-1 expression and endothelial dysfunction. Furthermore, these studies provide novel evidence that p65 levels were increased in SS mouse lungs and HEM-treated HPAECs and that p65 loss of function was sufficient to attenuate HEM-induced increases in HPAEC ET-1 expression. These results suggest that targeting PPARγ-HUWE1-miR-98 signaling may represent a novel therapeutic approach in SCD-PH pathogenesis.
Requests for data sharing should be e-mailed to the corresponding author, Bum-Yong Kang (bum-yong.kang@emory.edu).
Acknowledgments
The authors thank Victor Tseng for his careful review and valuable editorial comments in preparation of the manuscript.
This study was supported by funding from the National Institutes of Health, National Heart, Lung, and Blood Institute (grants R01 HL102167 [C.M.H. and R.L.S.], R01 HL119291 [C.P.], and R01 HL133053 [B.-Y.K.]), the Center for Drug Discovery (grant CDD-0075064) (R.L.S. and B.-Y.K.), Veterans Affairs Biomedical Laboratory Research and Development (VA BLR&D) Merit Review Award (I01 BX004263) (C.M.H.), and Emory/Childrens Healthcare CEB (grant F16788-00) (B.-Y.K.).
The contents of this article do not represent the views of the Department of Veterans Affairs or the US Government.
Authorship
Contribution: B.-Y.K. created the concept, hypothesis delineation, and design; A.J.J., S.S.C., C.P., C.-M.L., R.L.B., M.J.P., J.M., D.R.A., R.L.S., C.M.H., and B.-Y.K. acquired, analyzed, and interpreted the data; and A.J.J., S.S.C., C.M.H., and B.-Y.K. wrote the article.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Bum-Yong Kang, Division of Pulmonary, Allergy, Critical Care and Sleep Medicine, Atlanta Veterans Affairs Healthcare, 1670 Clairmont Rd, Decatur, GA 30033; e-mail: bum-yong.kang@emory.edu.

