

Key Points
AZA depletes HSCs and enables donor engraftment in immunocompetent mice.
Anti-CD117 mAb in combination with AZA significantly enhances HSC depletion and donor engraftment in immunocompetent mice.

Abstract
Depletion of hematopoietic stem cells (HSCs) is used therapeutically in many malignant and nonmalignant blood disorders in the setting of a hematopoietic cell transplantation (HCT) to eradicate diseased HSCs, thus allowing donor HSCs to engraft. Current treatments to eliminate HSCs rely on modalities that cause DNA strand breakage (ie, alkylators, radiation) resulting in multiple short-term and long-term toxicities and sometimes even death. These risks have severely limited the use of HCT to patients with few to no comorbidities and excluded many others with diseases that could be cured with an HCT. 5-Azacytidine (AZA) is a widely used hypomethylating agent that is thought to preferentially target leukemic cells in myeloid malignancies. Here, we reveal a previously unknown effect of AZA on HSCs. We show that AZA induces early HSC proliferation in vivo and exerts a direct cytotoxic effect on proliferating HSCs in vitro. When used to pretreat recipient mice for transplantation, AZA permitted low-level donor HSC engraftment. Moreover, by combining AZA with a monoclonal antibody (mAb) targeting CD117 (c-Kit) (a molecule expressed on HSCs), more robust HSC depletion and substantially higher levels of multilineage donor cell engraftment were achieved in immunocompetent mice. The enhanced effectiveness of this combined regimen correlated with increased apoptotic cell death in hematopoietic stem and progenitor cells. Together, these findings highlight a previously unknown therapeutic mechanism for AZA which may broaden its use in clinical practice. Moreover, the synergy we show between AZA and anti-CD117 mAb is a novel strategy to eradicate abnormal HSCs that can be rapidly tested in the clinical setting.
Introduction
Eradicating dysfunctional or disease-causing hematopoietic stem cells (HSCs) and replacing them with healthy HSCs is the curative strategy conferred by hematopoietic cell transplantation (HCT). A broad array of blood and immune disorders ranging from nonmalignant genetic disorders (eg, the hemoglobinopathies or immune deficiencies) to malignant clonal disorders (eg, acute leukemias) can be corrected by HCT. HSCs occupy specialized marrow niches which support their unique properties of self-renewal and maintenance, and host HSCs must be actively eradicated to permit donor HSC engraftment. Currently, HCT continues to depend on DNA-damaging alkylators (ie, busulfan, melphalan) or radiation to achieve eradication of HSCs and clearing of marrow niche space. The development of specific and less toxic preparative regimens would substantially reduce the morbidity and mortality of HCT, thereby expanding the scope of the diseases treated and allowing many more patients to undergo this life-saving procedure which is currently considered to be too high risk.1-4
To overcome this problem of HCT, we have pursued development of targeted HSC depletion using a monoclonal antibody (mAb) that recognizes CD117 (c-Kit), a receptor tyrosine kinase present on hematopoietic stem and progenitor cells (HSPCs), and showed that the anti-human CD117 mAb JSP191 (formerly AMG191) can eliminate HSCs with little to no off-target toxicity in non-human primates.5 Furthermore, a phase 1 clinical trial using JSP191 as the sole conditioning agent for patients undergoing HCT for severe combined immunodeficiency (SCID) has demonstrated that the antibody can safely clear HSC niche space, thus permitting donor HSC engraftment and, as a result, reconstituting robust T-cell immunity.6,7 CD117 is also expressed on the clonally abnormal disease-initiating HSCs that underlie the cause of myelodysplastic syndrome (MDS). We showed that in mice xenografted with human MDS HSCs, anti-human CD117 mAbs can deplete these cells.8 Hence, the effects of anti-CD117 mAbs on both nonmalignant and malignant dysfunctional HSCs make this biologic therapeutic agent suitable for different blood diseases ranging from monogenic disorders such as the hemoglobinopathies to bone marrow (BM) clonal malignant diseases. Because resistance to HSC depletion varies among the different disease states, we sought to enhance the potency of naked anti-CD117 mAbs. To date, strategies shown to enhance the capacity of anti-CD117 mAbs to mediate HSC ablation include the generation of antibody drug conjugates or combining anti-CD117 mAbs with low-dose irradiation or with agents that block the macrophage checkpoint inhibitor CD47.9-11 Here, we show that the widely used agent 5-Azacytidine (AZA) synergizes with ACK2 to deplete normal HSCs and enable donor HSC engraftment in wild-type mice.
AZA is a nucleoside analog best known as an epigenetic modifier used for treating MDS,12,13 acute myeloid leukemia (AML),14 and chronic myelomonocytic leukemia (CMML).15,16 AZA is given in multiple repeating cycles and is generally well tolerated and administered to patients who, because of advanced age or comorbidities, are not eligible for conventional chemotherapies. The antitumor activity of AZA is not fully understood but has been attributed to multiple mechanisms beyond re-expression of silenced tumor suppressor genes, such as direct apoptotic effects on malignant cells,17 immune-mediated response against tumor cells,18 differentiation induction,19,20 and effects on the BM microenvironment.21 Although myelosuppression is the most common hematologic adverse event,12,13 little is known about the effects of AZA on hematopoiesis in vivo.
Here we show, for the first time, that AZA causes depletion of short-term HSCs (ST-HSCs) and long-term HSCs (LT-HSCs) in the BM, sufficient to permit low-level engraftment of transplanted donor HSCs in wild-type mice. Moreover, when combined with the anti-CD117 mAb ACK2, AZA caused marked and prolonged depletion of recipient HSCs and resulted in robust levels of donor HSC engraftment in immunocompetent mice. The revelation that AZA alone has HSC depleting activity, and that this effect can be amplified by safe and targeted HSC depletion using an anti-CD117 mAb, promises to quickly translate into new therapeutic strategies to eradicate diseased HSCs.
Materials and methods
Mice
C57BL/6 (B6) mice (H-2b, Thy1.1, CD45.1/CD45.2) were recipients, and B6 mice (H-2b, Thy1.1, CD45.1) were donors for the congenic transplantation experiments. BALB.B mice (H-2b, Thy1.2, CD45.2) were recipients, and B6 mice (H-2b, Thy1.1, CD45.1) were donors for experiments in minor histocompatibility complex– mismatched transplantation (MHC-matched). Transplant recipients were >8 weeks old and donors were 8 to 12 weeks old. B6 (H-2b, Thy1.1, CD45.2) mice were used for in vivo depletion studies. Mice were bred and maintained under pathogen-free conditions at the Stanford University Research Animal Facility. All experiments were performed under a protocol approved by the Stanford Administrative Panel on Laboratory Animal Care.
In vivo administration of therapeutic agents and cytokines
Anti-mouse CD117 mAb clone ACK2 (Bio X Cell) and clone 2B8 (Bio X Cell ) were injected retro-orbitally as a single dose of 500 µg after intraperitoneal administration of diphenhydramine (Benadryl). Anti-CD4 (clone GK1.5, Bio X Cell) and anti-CD8 (clone YTS169.4, Bio X Cell) were administered intraperitoneally at a dose of 100 µg in 100 µL of phosphate-buffered saline (PBS). AZA (STEMCELL Technologies) was reconstituted in PBS to a concentration of 5 mg/mL and kept at −20°C for up to 1 month. Before each injection, AZA was thawed at room temperature (RT), resuspended in PBS, and administrated intraperitoneally at 100 µL. Recombinant murine stem cell factor (SCF; PeproTech) was dissolved in deionized water to a concentration of 10 µg/mL and injected intraperitoneally at 1 µg/day.22
Cell harvest and flow cytometry
BM (hips, femurs, tibia, spine) and spleen were harvested from euthanized mice. Bones were crushed in PBS plus 2% bovine calf serum and filtered through a 70-µm filter (Falcon). Spleens were mashed directly in a 70-µm filter. Red blood cell lysis was performed with ACK lysis buffer for 10 minutes at RT. Cells were counted on a Countess automated cell counter (Invitrogen) and stained for 20 minutes at RT. Detailed information regarding antibodies and Ki67 extracellular staining is provided in the supplemental Data. Data were collected by using a BD FACSAria II flow cytometry system and analyzed with FlowJo software (version 9.9.4.). Absolute cell counts were calculated by multiplying the frequency of the population (determined by fluorescence-activated cell sorting [FACS]) by the total live BM cells.
HSC transplantation and chimerism analysis
BM cells were prepared as described above. For HSC transplantation, lineage depletion was performed with magnetic column separation and direct lineage cell depletion kit (MACS Separation Columns LS; Miltenyi Biotec) per the manufacturer’s instructions. HSCs (Lin–Sca1+c-Kit+ [LSK]) were sorted on the BD FACSAria II. Propidium iodide was used to exclude dead cells. BM or Lin–Sca1+c-Kit+ cells were resuspended in 100 µL PBS plus 2% fetal bovine serum and injected retro-orbitally into recipients. Multilineage donor chimerism was assessed in the peripheral blood (PB) every 4 to 6 weeks. Then, 50 to 100 µL of PB was collected via the tail vein, incubated with 2% dextran plus 10 mM EDTA in PBS, and left for 45 to 60 minutes at RT. Supernatant was extracted and red blood cells were lysed for 7 minutes at RT. Cells were stained and analyzed using the BD FACSAria II.
In vitro cell culture, imaging, and viability assay
Detailed description of the methods are provided in the supplemental Data. Briefly, FACS-purified mouse or human HSCs were cultured in the presence of SCF and thrombopoietin with or without the indicated concentrations of AZA. Cell imaging, counting, and toxicity assays were performed by ImagExpress Pico.
Statistical analysis
Mann-Whitney U tests were performed using GraphPad Prism software (version 8.4.2.). P values <.05 were reported as statistically significant. Kaplan-Meier survival curves were analyzed by log-rank tests.
Results
AZA depletes mature cells and HSPCs
We studied the effect of AZA on normal hematopoiesis and determined that AZA causes depletion of both mature cells and HSPCs. B6 mice were treated with 5 mg/kg per day AZA (approximately equivalent to the human dose of 75 mg/m2)23,24 for 5 days, and the blood, spleen, and BM were analyzed by extended phenotype on days 6, 10, 15, and 20 (Figure 1A). Overall BM cellularity was decreased on day 6 (80% from baseline; P = .0286; Figure 1B-C), and it recovered by day 20. In blood, there was transient reduction in all lineages except platelets, with the lowest values documented on day 10 (Figure 1D). Granulocytes were the most affected and showed an approximate fourfold reduction. All blood subsets recovered to normal ranges by day 20. Splenic myeloid cells were also significantly reduced on day 6, but they recovered by day 10. Splenic lymphocyte populations were only slightly affected by AZA (supplemental Figure 1).
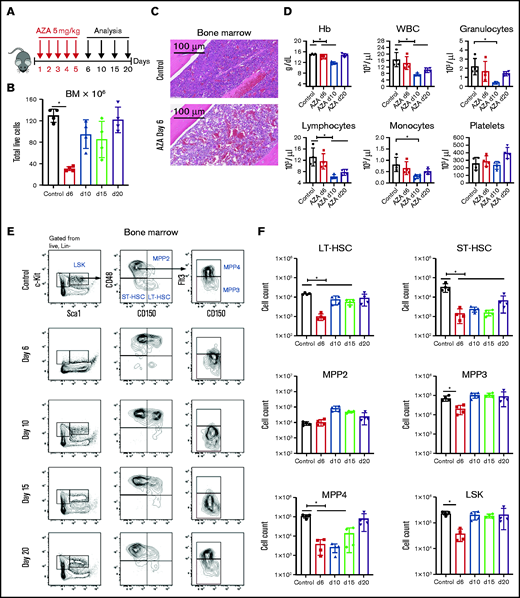
AZA depletes HSPCs in vivo. (A) Treatment schema. C57BL/6 mice were injected intraperitoneally with AZA at 5 mg/kg per day for 5 consecutive days. PB, spleens, and BM from both legs and spine were analyzed on days 6, 10, 15, and 20 after the start of AZA treatment. (B) Total live cells in the BM of untreated mice and AZA-treated mice on days 6, 10, 15, and 20 after the start of AZA treatment. (C) Hematoxylin and eosin staining of a BM section from 1 mouse femur on day 6 after treatment with AZA 5 mg/kg per day for 5 days compared with an untreated control. (D) PB cell counts for untreated mice compared with AZA-treated mice on days 6, 10, and 20 after the start of AZA treatment. (E) Representative flow cytometry contour plots of the HSPC compartment in the BM of untreated and AZA-treated mice on days 6, 10, 15, and 20. The figure shows our gating strategy beginning with Lin– live cells, LSK cells, MPPs, LT-HSCs, and ST-HSCs. (F) Absolute cell counts from the different HSPC compartments on days 6, 10, 15, and 20 after AZA treatment compared with untreated control mice. LSK: Lin–Sca1+c-Kit+; LT-HSC: LSKCD150+CD48–; ST-HSC: LSKCD150–CD48–; MPP2: LSKCD150+CD48+; MPP3: LSKCD150–CD48+Flt3–; MPP4: LSKCD150–CD48+Flt3+. Data are expressed as mean ± standard deviation (SD); n = 4 mice per group per time point. *P < .05. Hb, hemoglobin; WBC, white blood cell.
AZA depletes HSPCs in vivo. (A) Treatment schema. C57BL/6 mice were injected intraperitoneally with AZA at 5 mg/kg per day for 5 consecutive days. PB, spleens, and BM from both legs and spine were analyzed on days 6, 10, 15, and 20 after the start of AZA treatment. (B) Total live cells in the BM of untreated mice and AZA-treated mice on days 6, 10, 15, and 20 after the start of AZA treatment. (C) Hematoxylin and eosin staining of a BM section from 1 mouse femur on day 6 after treatment with AZA 5 mg/kg per day for 5 days compared with an untreated control. (D) PB cell counts for untreated mice compared with AZA-treated mice on days 6, 10, and 20 after the start of AZA treatment. (E) Representative flow cytometry contour plots of the HSPC compartment in the BM of untreated and AZA-treated mice on days 6, 10, 15, and 20. The figure shows our gating strategy beginning with Lin– live cells, LSK cells, MPPs, LT-HSCs, and ST-HSCs. (F) Absolute cell counts from the different HSPC compartments on days 6, 10, 15, and 20 after AZA treatment compared with untreated control mice. LSK: Lin–Sca1+c-Kit+; LT-HSC: LSKCD150+CD48–; ST-HSC: LSKCD150–CD48–; MPP2: LSKCD150+CD48+; MPP3: LSKCD150–CD48+Flt3–; MPP4: LSKCD150–CD48+Flt3+. Data are expressed as mean ± standard deviation (SD); n = 4 mice per group per time point. *P < .05. Hb, hemoglobin; WBC, white blood cell.
In contrast to the relatively mild effects on the peripheral hematopoietic compartments, analyses of BM subpopulations showed that AZA caused marked depletion of all CD117+ cells (Figure 1E). Both LT-HSCs (Lin–Sca1+c-Kit+CD150+CD48–) and ST-HSCs (LSKCD150–CD48–) were markedly reduced, with nadirs on day 6 at 15-fold and 24-fold, respectively. Recovery of ST-HSCs was delayed beyond day 20 (Figure 1F). Examination of splenic populations did not suggest that mobilization of HSCs to secondary organs caused their decrease in BM (supplemental Figure 2). Rather, splenic ST- and LT-HSCs were also reduced on day 6 with rebound of LT-HSCs by day 10. We also performed titration studies of AZA. A dose of 1.25 mg/kg per day resulted in an ∼25% depletion of HSPCs and LT-HSCs (supplemental Figure 3A-B) in BM, but this reduction was markedly less than the depletion caused by 2.5 and 5.0 mg/kg per day.
In examining the kinetics of depletion and recovery of subpopulations within the BM, we noted that depletion of the myeloid progenitors (MPs), including common MPs, granulocyte-macrophage progenitors, and megakaryocyte-erythrocyte progenitors (supplemental Figure 4A-B) was followed by rapid expansion of myeloid-biased multipotent progenitors (MPPs) defined phenotypically as LSKCD150+CD48+ (MPP2) (Figure 1F). In parallel, there was a proportional increase by day 6 in both MPP2 and MPP3 (LSKCD150–CD48+Flt3–) cells (supplemental Figure 5). The pattern of depletion and recovery differed for the lymphoid-biased MPP4 (LSKCD150–CD48+Flt3+) progenitors reaching nadirs on day 10 with slower recovery by day 20 (Figure 1F). Another notable effect of AZA was marked and prolonged depletion of BM B cells measured by the CD19 marker. Maximal B-cell depletion of >30-fold occurred at day 10 after AZA treatment and did not recover until >day 15 (supplemental Figure 6). BM natural killer cells also transiently declined, whereas CD8+ T cells transiently increased, normalizing by day 10.
AZA-induced HSC proliferation in vivo correlates with direct toxic effects on HSCs
In vivo depletion of HSCs by AZA has not been previously reported and was surprising because most HSCs are quiescent, and nonproliferating cells are known to be insensitive to the toxicities of AZA.25 Given the rapid recovery of mature myeloid cells and the shift of hematopoiesis toward the more actively cycling myeloid-biased MPP2/MPP3 fractions, we hypothesized that AZA’s depletion of mature cells might drive proliferation of dormant HSCs thereby increasing their sensitivity to the effects of AZA effects in vivo. Thus, to assess the proliferative state of HSPCs after AZA treatment, mice received AZA 5 mg/kg per day for 3 days, and Ki67 protein expression of HSPCs and MPs (Lin–Sca1–c-Kit+) was assessed on day 4. We observed significant increase of Ki67 in both MPs and HSPCs, including LT-HSCs (supplemental Figure 7A-B). To confirm that HSCs are actively entering the S-phase of the cell cycle, which could permit AZA incorporation into DNA, we performed in vivo analysis of bromodeoxyuridine (BrdU) incorporation. Mice were treated for 3 days with AZA 2.5 mg/kg per day or 5 mg/kg per day, and after the last AZA dose, BrdU was administered as previously described26 (Figure 2A). Significant increase in BrdU incorporation was observed in the LT-HSCs that had been treated with AZA compared with untreated controls (Figure 2B-C).
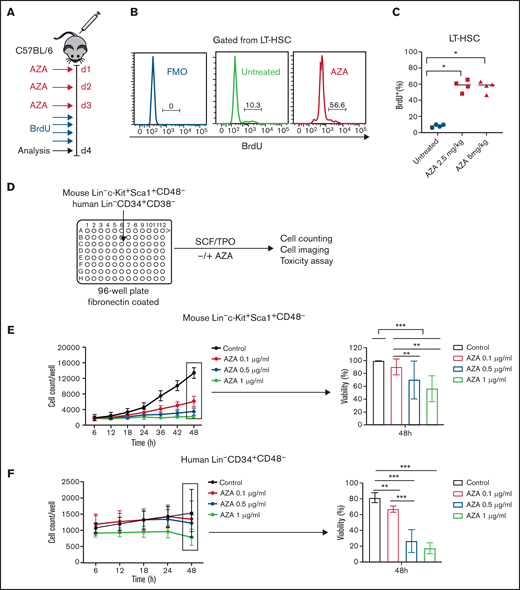
AZA induces proliferation of HSCs in vivo and reduces growth and viability of proliferating HSCs in vitro. (A) Treatment schema. C57BL/6 mice were injected intraperitoneally with AZA at 2.5 or 5 mg/kg per day for 3 days. After the last AZA dose, BrdU was administrated intraperitoneally at 1 mg per mouse once every 6 hours for a total of 24 hours. Mice were euthanized on day 4 after the start of experiment, and BM from both legs was analyzed by flow cytometry for incorporation of BrdU. (B) Representative histogram of BrdU expression in LT-HSCs (gated from live Lin–Sca1+c-Kit+CD150+CD48– cells) from 1 untreated and 1 AZA-treated mouse BM sample. (C) BrdU expression in percent for LT-HSCs (n = 4 mice per group). (D) Schematic of in vitro culture protocol. FACS-sorted mouse Lin–Sca1+c-Kit+CD48– or human Lin–CD34+CD38– cells were plated in 96-well flat-bottom plates coated with fibronectin that contained 100 µL of HSC media per well, and were supplemented with 10 ng/mL SCF and 100 ng/mL thrombopoietin (TPO). Different concentrations of AZA (0.1, 0.5, and 1 µg/mL) were added on 2 consecutive days (at baseline and at 24 hours after cell culturing). Cell counting and cell imaging was performed once every 6 hours for a total of 48 hours on an ImagExpress Pico automated cell counting system. After 48 hours, the percentage of live and dead cells was assessed by using EarlyTox Live/Dead Assay Kit. (E) Proliferation curves of FACS-sorted mouse Lin–Sca1+c-Kit+CD48– cells in the presence of indicated concentrations of AZA (left). Cell viability was assessed by the percentage of calcein AM+ cells after 48 hours of cell culture (right) (data are from 3 independent experiments). (F) Proliferation curves of FACS-sorted human Lin–CD34+CD38– cells in the presence of indicated concentrations of AZA (left). Cell viability was assessed by the percentage of calcein AM+ cells after 48 hours of cell culture (right) (data are from 2 independent experiments). Data are expressed as mean ± SD. *P < .05; **P < .01; ***P < .001.
AZA induces proliferation of HSCs in vivo and reduces growth and viability of proliferating HSCs in vitro. (A) Treatment schema. C57BL/6 mice were injected intraperitoneally with AZA at 2.5 or 5 mg/kg per day for 3 days. After the last AZA dose, BrdU was administrated intraperitoneally at 1 mg per mouse once every 6 hours for a total of 24 hours. Mice were euthanized on day 4 after the start of experiment, and BM from both legs was analyzed by flow cytometry for incorporation of BrdU. (B) Representative histogram of BrdU expression in LT-HSCs (gated from live Lin–Sca1+c-Kit+CD150+CD48– cells) from 1 untreated and 1 AZA-treated mouse BM sample. (C) BrdU expression in percent for LT-HSCs (n = 4 mice per group). (D) Schematic of in vitro culture protocol. FACS-sorted mouse Lin–Sca1+c-Kit+CD48– or human Lin–CD34+CD38– cells were plated in 96-well flat-bottom plates coated with fibronectin that contained 100 µL of HSC media per well, and were supplemented with 10 ng/mL SCF and 100 ng/mL thrombopoietin (TPO). Different concentrations of AZA (0.1, 0.5, and 1 µg/mL) were added on 2 consecutive days (at baseline and at 24 hours after cell culturing). Cell counting and cell imaging was performed once every 6 hours for a total of 48 hours on an ImagExpress Pico automated cell counting system. After 48 hours, the percentage of live and dead cells was assessed by using EarlyTox Live/Dead Assay Kit. (E) Proliferation curves of FACS-sorted mouse Lin–Sca1+c-Kit+CD48– cells in the presence of indicated concentrations of AZA (left). Cell viability was assessed by the percentage of calcein AM+ cells after 48 hours of cell culture (right) (data are from 3 independent experiments). (F) Proliferation curves of FACS-sorted human Lin–CD34+CD38– cells in the presence of indicated concentrations of AZA (left). Cell viability was assessed by the percentage of calcein AM+ cells after 48 hours of cell culture (right) (data are from 2 independent experiments). Data are expressed as mean ± SD. *P < .05; **P < .01; ***P < .001.
To directly determine the effects of AZA on the survival of proliferating mouse and human HSCs, we performed in vitro toxicity studies. FACS-purified mouse LSKCD48– or human Lin–CD34+CD38– cells (containing both LT- and ST-HSCs) were cultured in the presence of SCF and thrombopoietin plus AZA at concentrations of 0.1, 0.5, and 1 µg/mL added to the cultures on 2 consecutive days (at baseline and after 24 hours). The concentration range was chosen based upon the plasma concentrations documented in patients after subcutaneous administration of the standard 75 mg/m2 AZA (Cmax = 750 ± 403.3 ng/mL).27 Cell imaging and counting was performed every 6 hours, and viability was measured at 48 hours by using the ImagExpress Pico automated cell counting system (Figure 2D). In both mouse and human HSC cultures, AZA reduced the number of cells in a dose-dependent manner and showed significant decrease in cell viability as assessed by calcein AM (Figure 2E-F).
In vivo direct killing by apoptosis was confirmed by flow cytometry using annexin V and propidium iodide staining of the HSPC fractions. Increases in apoptotic cell death in HSPCs was detectable after 3 days of AZA treatment and was dose dependent (supplemental Figure 8A-B). Collectively, these data suggest that AZA induces HSC proliferation in vivo, which increases the susceptibility of this population to the direct toxic effects of the drug.
Anti-CD117 mAb ACK2 combined with AZA augments HSC depletion
HSC survival is known to depend on SCF-CD117 signaling,28,29 and it was previously shown that SCF-neutralizing antibodies can mitigate hematopoietic recovery in the setting of hematopoietic injury after sublethal total body irradiation.30 On the basis of our observation that AZA is directly cytotoxic to proliferating HSCs in vitro and induces apoptotic cell death of HSPCs in vivo, we hypothesized that pharmacologic blockade of SCF-CD117 signaling with the anti-mouse anti-CD117 mAb ACK2 would enhance HSC depletion by increasing apoptotic cell death by blocking an important survival signaling for HSCs. Although all HSPCs express c-Kit, it is known that primarily HSCs with long-term repopulating potential depend on SCF-CD117 signaling for their survival.28,29 ACK2 was given as a single dose of 500 µg 5 days before treatment with AZA, as shown in Figure 3A. This combination treatment resulted in marked reduction in overall BM cellularity (Figure 3B) and robust depletion of HSPCs (Figure 3C). The depth of LT-HSC depletion and time to recovery was significantly greater than with treatment with AZA alone at all time points, and recovery of ST-HSCs was also delayed in the ACK2-AZA group (Figure 3D). MPPs were less affected by the ACK2-AZA combination compared with the AZA alone group, suggesting that the addition of ACK2 to AZA mainly affected the HSC populations. Splenic HSCs were also reduced, which suggests that HSC depletion was not a result of mobilization (supplemental Figure 9). LSK cells from mice treated with ACK2-AZA (2.5 mg/kg for 3 days; Figure 3G) demonstrated significant increase in apoptotic cell death as assessed by annexin V and propidium iodide staining compared with mice treated with either agent alone (Figure 3H-I). In addition, treatment with ACK2-AZA also caused marked but transient decline of all PB cell counts (supplemental Figure 10A,C). Because of this steep decline in hemoglobin, these mice were either transfused to correct the anemia or observed without intervention. Both groups had 100% survival (supplemental Figure 10B), demonstrating that ACK2-AZA did not cause a long-lasting lethal pancytopenia.
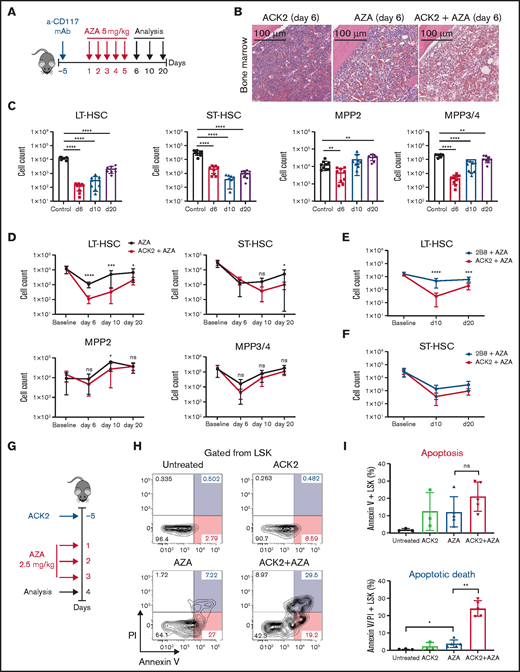
Anti-CD117 mAb (ACK2) combined with AZA enhances HSC depletion and delays HSC recovery in vivo. (A) Treatment schema for panels B-F. C57BL/6 mice were injected intravenously with a single dose of ACK2 or 2B8 at 500 µg 5 days before the start of treatment with AZA. AZA was administered intraperitoneally at 5 mg/kg once per day for 5 days. Mice were euthanized at 6, 10, or 20 days after the first dose of AZA; spleens and BM from both legs and spine were harvested and analyzed by flow cytometry for HSC depletion and depletion of mature myeloid and lymphoid cells. (B) Hematoxylin and eosin staining of a BM section of 1 femur at day 6 from a mouse treated with ACK2 only, AZA only, or ACK2-AZA. (C) Absolute cell counts of the different HSPC compartments in the BM of untreated controls and ACK2-AZA–treated mice on days 6, 10, and 20 after the start of AZA treatment as measured by flow cytometry. (D) Comparison of the absolute cell counts of different HSPC compartments in the BM of mice treated with AZA only or ACK2-AZA at baseline and at 6, 10, and 20 days after start of AZA. (E) Comparison of the absolute cell counts of LT-HSCs in the BM of mice treated with 2B8-AZA vs ACK2-AZA at baseline and on days 10 and 20 after the start of AZA treatment. (F) Comparison of the absolute cell counts of ST-HSCs in the BM of mice treated with 2B8-AZA vs ACK2-AZA at baseline and on days 10 and 20 after the start of AZA treatment. For panels C-F, data for each experimental group were pooled from 2 independent experiments; n = 8-9 mice per group per time point. (G) Treatment schema for panels H-I. C57BL/6 mice were injected intravenously with a single dose of ACK2 500 µg 5 days before the start of treatment with AZA (administered intraperitoneally at a dose of 2.5 mg/kg per day for 3 days). Mice were euthanized 4 days after the first dose of AZA, and BM from both legs was harvested and analyzed by flow cytometry for annexin V and propidium iodide (PI) staining. (H) Representative flow cytometry plots of annexin V and PI staining (gated from Lin–Sca1+c-Kit+) in untreated controls and mice treated with ACK2, AZA 2.5 mg/kg, or ACK2-AZA. (I) Top: annexin V+; bottom: annexin V+/PI+; stained cells are shown as a percentage of LSK cells in the different treatment groups compared with untreated controls (n = 3-5 mice). Data are expressed as mean ± SD. *P < .05; **P < .01; ***P < .001; ****P < .0001. ns, nonsignificant.
Anti-CD117 mAb (ACK2) combined with AZA enhances HSC depletion and delays HSC recovery in vivo. (A) Treatment schema for panels B-F. C57BL/6 mice were injected intravenously with a single dose of ACK2 or 2B8 at 500 µg 5 days before the start of treatment with AZA. AZA was administered intraperitoneally at 5 mg/kg once per day for 5 days. Mice were euthanized at 6, 10, or 20 days after the first dose of AZA; spleens and BM from both legs and spine were harvested and analyzed by flow cytometry for HSC depletion and depletion of mature myeloid and lymphoid cells. (B) Hematoxylin and eosin staining of a BM section of 1 femur at day 6 from a mouse treated with ACK2 only, AZA only, or ACK2-AZA. (C) Absolute cell counts of the different HSPC compartments in the BM of untreated controls and ACK2-AZA–treated mice on days 6, 10, and 20 after the start of AZA treatment as measured by flow cytometry. (D) Comparison of the absolute cell counts of different HSPC compartments in the BM of mice treated with AZA only or ACK2-AZA at baseline and at 6, 10, and 20 days after start of AZA. (E) Comparison of the absolute cell counts of LT-HSCs in the BM of mice treated with 2B8-AZA vs ACK2-AZA at baseline and on days 10 and 20 after the start of AZA treatment. (F) Comparison of the absolute cell counts of ST-HSCs in the BM of mice treated with 2B8-AZA vs ACK2-AZA at baseline and on days 10 and 20 after the start of AZA treatment. For panels C-F, data for each experimental group were pooled from 2 independent experiments; n = 8-9 mice per group per time point. (G) Treatment schema for panels H-I. C57BL/6 mice were injected intravenously with a single dose of ACK2 500 µg 5 days before the start of treatment with AZA (administered intraperitoneally at a dose of 2.5 mg/kg per day for 3 days). Mice were euthanized 4 days after the first dose of AZA, and BM from both legs was harvested and analyzed by flow cytometry for annexin V and propidium iodide (PI) staining. (H) Representative flow cytometry plots of annexin V and PI staining (gated from Lin–Sca1+c-Kit+) in untreated controls and mice treated with ACK2, AZA 2.5 mg/kg, or ACK2-AZA. (I) Top: annexin V+; bottom: annexin V+/PI+; stained cells are shown as a percentage of LSK cells in the different treatment groups compared with untreated controls (n = 3-5 mice). Data are expressed as mean ± SD. *P < .05; **P < .01; ***P < .001; ****P < .0001. ns, nonsignificant.
To further interrogate whether CD117 blockade is needed to augment HSC depletion by AZA, we tested another anti-CD117 antibody, clone 2B8, which is known to only partially inhibit SCF-driven hematopoiesis in vitro compared with ACK2.31 Treatment of mice with 2B8-AZA did not achieve the same depth or duration of HSC depletion as ACK2-AZA (Figure 3E-F) which led us to conclude that ACK2 disruption of SCF-CD117 signaling exacerbates the effects of AZA on depletion of HSCs, and that excess SCF might be able to mitigate this effect. To test this idea, we treated mice with ACK2-AZA plus 1 µg of exogenous murine SCF for 5 consecutive days (supplemental Figure 11A). Mice treated with SCF demonstrated significantly faster recovery of LT-HSCs in the BM at day 11, with no changes in the recovery of ST-HSCs, MPP2, MPP3, and MPP4 cells (supplemental Figure 11B). The same trend was observed in the spleens of treated mice, although the kinetics of LT-HSC recovery did not reach statistical significance (supplemental Figure 11C). Taken together, these data support the idea that the SCF-CD117 interaction plays an important role in HSPC restoration after treatment with AZA.
ACK2 combined with AZA enables engraftment of congenic HSCs
We next tested treatment with ACK2-AZA to determine whether this combined regimen would enable donor HSC engraftment in immunocompetent mice. We applied the same treatment protocol described in the depletion studies above. Conditioning with ACK2 was given 5 days before treatment with AZA, and AZA was administrated for 5 days, followed by transplantation of 20 × 106 congenic whole BM cells or 5 × 104 LSK cells (Figure 4A). Recipients were observed by using analyses of blood donor chimerism as assessed by CD45 markers for the different cell populations (supplemental Figure 12) up to week 24. Sustained multilineage donor chimerism was achieved with transplantation of whole BM cells using ACK2 plus 5 days of treatment with AZA at 2.5 mg/kg (Figure 4B), whereas neither ACK2 treatment alone nor 5 days of treatment with AZA 2.5 mg/kg alone resulted in stable engraftment. Conditioning with ACK2 and AZA at a higher dose of 5 mg/kg led to significantly increased stable multilineage donor chimerism levels (>50%; Figure 4B). Interestingly, single-agent AZA at 5 mg/kg also enabled sustained donor engraftment, albeit at lower levels of myeloid donor chimerism of 10% to 15%, underscoring the degree to which AZA itself depletes HSCs. Similar synergistic activity between ACK2 and AZA was achieved when purified congenic LSK HSCs were transplanted into mice conditioned with ACK2-AZA at the 5 mg/kg dose. The median level of long-term myeloid chimerism, which correlates with HSC engraftment31 in this group, was 27% (Figure 4C). Of note, in performing these experiments, we observed that concomitant administration of ACK2 and AZA or administration of ACK2 after AZA was not effective in permitting enhanced HSC donor engraftment compared with AZA alone (data not shown). When these data are taken together, we can conclude that treatment with the ACK2-AZA combination led to enhanced and sustained levels of donor chimerism and that these synergistic effects depend on the timing of anti-CD117 administration because SCF-CD117 blockade should precede AZA treatment.
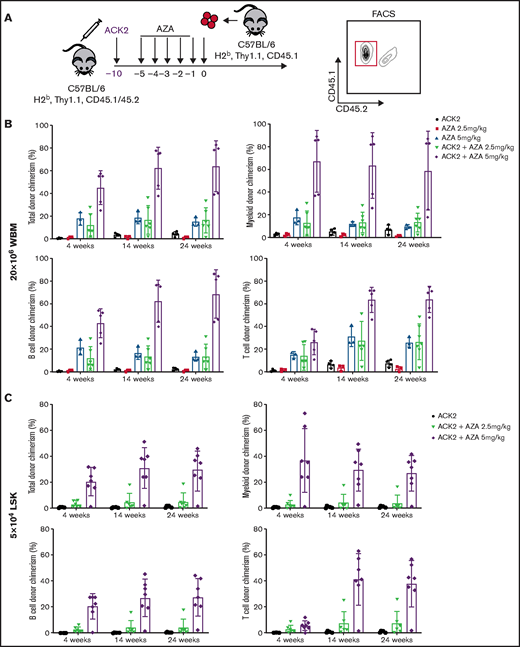
ACK2 synergizes with AZA and permits engraftment of congenic HSCs in immunocompetent mice. (A) Schematic of congenic transplantations. C57BL/6 (H-2b, Thy1.1, CD45.1) mice were donors for C57BL/6 (H-2b, Thy1.1, CD45.1/CD45.2) recipients. ACK2 at 500 µg was injected intravenously 10 days before cell infusion, and AZA was administrated on days −5 through −1 at a dose of 2.5 or 5 mg/kg per day. Recipients received 20 × 106 whole BM cells (WBM) or 5 × 104 FACS-sorted LSK cells on day 0 via retro-orbital injection. PB chimerism was assessed by flow cytometry using the CD45 allelic marker to distinguish between donor and recipient live total cells, myeloid cells (Gr1+Mac1+), B cells (CD19+CD3–), and T cells (CD19–CD3+). (B) Higher levels of sustained multilineage donor engraftment were observed in the ACK2-AZA group compared with single-agent ACK2 or single-agent AZA groups after transplantation of 20 × 106 WBM cells (n = 3-6 mice per group). (C) ACK2-AZA at 5 mg/kg per day enables sustained multilineage engraftment of 5 × 104 FACS donor congenic LSK cells (n = 6-7 mice per group). Data were pooled from 2 independent experiments and represent mean ± SD.
ACK2 synergizes with AZA and permits engraftment of congenic HSCs in immunocompetent mice. (A) Schematic of congenic transplantations. C57BL/6 (H-2b, Thy1.1, CD45.1) mice were donors for C57BL/6 (H-2b, Thy1.1, CD45.1/CD45.2) recipients. ACK2 at 500 µg was injected intravenously 10 days before cell infusion, and AZA was administrated on days −5 through −1 at a dose of 2.5 or 5 mg/kg per day. Recipients received 20 × 106 whole BM cells (WBM) or 5 × 104 FACS-sorted LSK cells on day 0 via retro-orbital injection. PB chimerism was assessed by flow cytometry using the CD45 allelic marker to distinguish between donor and recipient live total cells, myeloid cells (Gr1+Mac1+), B cells (CD19+CD3–), and T cells (CD19–CD3+). (B) Higher levels of sustained multilineage donor engraftment were observed in the ACK2-AZA group compared with single-agent ACK2 or single-agent AZA groups after transplantation of 20 × 106 WBM cells (n = 3-6 mice per group). (C) ACK2-AZA at 5 mg/kg per day enables sustained multilineage engraftment of 5 × 104 FACS donor congenic LSK cells (n = 6-7 mice per group). Data were pooled from 2 independent experiments and represent mean ± SD.
ACK2 combined with AZA enables engraftment of allogeneic HSCs
Next, we tested whether ACK2-AZA conditioning could be applied to the allogeneic setting by performing HSC transplantations across minor histocompatibility complex barriers (Figure 5A). Then, 5 × 104 LSK cells purified from B6 donors (H-2b, Thy1.1, CD45.1) were transplanted into BALB.B recipients (H-2b, Thy1.2, CD45.2) after conditioning with ACK2, with 5 days of AZA 5 mg/kg, or with the combination. In this model, anti-CD4 and anti-CD8 mAbs were administrated for 3 consecutive days before donor cell infusion to minimize immune-mediated rejection of the allogeneic donor cells. Blood chimerism analyses were performed up to week 26 after HCT. Similar to engraftment in the congenic setting, successful sustained multilineage donor engraftment was achieved in the ACK2-AZA group (Figure 5B), whereas none to minimal engraftment was achieved when either agent alone was used as conditioning. In the ACK2-AZA group, the median long-term levels of myeloid and total chimerism were 32% and 40%, respectively. These data suggest that anti-CD117 mAb plus AZA can be considered a conditioning approach for allogeneic HCT.
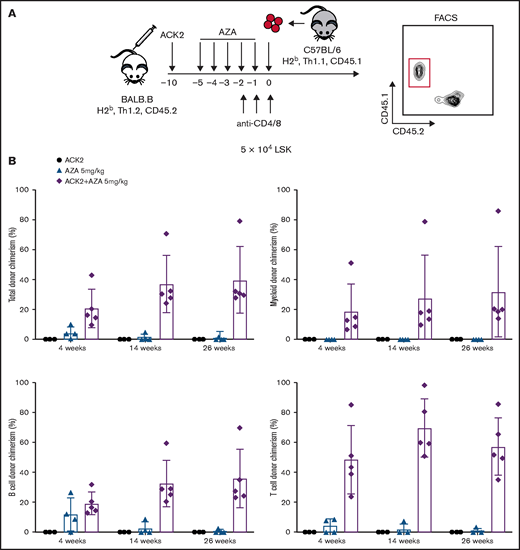
ACK2 synergizes with AZA and permits engraftment of allogeneic HSCs in immunocompetent mice. (A) Schematic of allogeneic transplantations. BALB.B mice (H-2b, Thy1.2, CD45.2) were recipients for B6 donors (H-2b, Thy1.1, CD45.1). ACK2 was injected intravenously 10 days before transplantation, AZA was administered intraperitoneally on days −5 through −1 at 5 mg/kg per day, and anti-CD4/anti-CD8 mAbs were injected intravenously at 100 µg for each mAb on days −2, −1, 0 (day of transplant). Recipients received 5 × 104 LSK cells at day 0 via retro-orbital injection. PB chimerism analysis was assessed by flow cytometry using a CD45 marker to distinguish between donor and recipient live total, myeloid (Gr1+Mac1+), B cells (CD19+CD3–), and T cells (CD19–CD3+). (B) Multilineage donor-derived chimerism in PB at 4, 14, and 26 weeks after conditioning with single-agent ACK2, single-agent AZA 5 mg/kg per day, or ACK2-AZA and transplantation of 5 × 104 LSK cells. Data represent mean ± SD (n = 3-5 mice per group).
ACK2 synergizes with AZA and permits engraftment of allogeneic HSCs in immunocompetent mice. (A) Schematic of allogeneic transplantations. BALB.B mice (H-2b, Thy1.2, CD45.2) were recipients for B6 donors (H-2b, Thy1.1, CD45.1). ACK2 was injected intravenously 10 days before transplantation, AZA was administered intraperitoneally on days −5 through −1 at 5 mg/kg per day, and anti-CD4/anti-CD8 mAbs were injected intravenously at 100 µg for each mAb on days −2, −1, 0 (day of transplant). Recipients received 5 × 104 LSK cells at day 0 via retro-orbital injection. PB chimerism analysis was assessed by flow cytometry using a CD45 marker to distinguish between donor and recipient live total, myeloid (Gr1+Mac1+), B cells (CD19+CD3–), and T cells (CD19–CD3+). (B) Multilineage donor-derived chimerism in PB at 4, 14, and 26 weeks after conditioning with single-agent ACK2, single-agent AZA 5 mg/kg per day, or ACK2-AZA and transplantation of 5 × 104 LSK cells. Data represent mean ± SD (n = 3-5 mice per group).
Discussion
These studies reveal for the first time that the hypomethylating agent AZA potently depletes HSCs, a finding that expands our knowledge of how this drug acts and opens the possibility for using AZA therapeutically for new indications. Specifically, we have leveraged this discovery to demonstrate that AZA combined with an antibody that targets the CD117 present on HSCs can be used as a novel HCT conditioning strategy and thereby replace highly genotoxic standard-of-care alkylating agents or radiation. Depletion of genetically aberrant or clonally abnormal HSCs is a requirement of all HCT conditioning regimens so they can achieve successful engraftment of donor allogeneic or autologous gene-corrected HSCs. Anti-CD117 antibodies have only recently reached the clinic6,7 as a new class of biologic agents capable of safely depleting HSCs. Their specificity, outstanding safety profile, and unique mechanism of action make these antibodies ideal for augmenting the activity of other HSC-depleting modalities. The possibility that the standard genotoxic modalities can be replaced by 2 much safer agents, AZA and anti-CD117 antibody, is a strategy that can be rapidly tested in clinical trials.
Our determination that AZA depletes HSCs and thus could be a therapeutic partner with anti-CD117 antibodies to augment the HSC engraftment was surprising. Hypomethylating agents have not been previously used as components of conditioning for transplantation. Although these agents have been shown to suppress myelopoiesis in the setting of minimal or no measurable malignant disease32,33 (eg, after allogeneic HCT as maintenance therapy), the effect of these drugs on hematopoiesis in general has not been well studied. The depletion of HSCs by AZA alone was unexpected because the vast majority (>95%) of HSCs are noncycling,34 and hypomethylating agents are known to rely on incorporation of drug metabolites into DNA during S-phase to exert their effects.25,35 Direct toxicity reportedly occurs via irreversible binding of the incorporated drug metabolites to DNA methyltransferase (DNMT), and it is these DNMT-AZA adducts that are cytotoxic, rather than the hypomethylated state caused by this binding.36 Because cells with higher levels of DNMT are reported to be more susceptible to the toxic effects of hypomethylating agents and because HSCs have been shown to have high levels of maintenance DNMT1 and high levels of DNA methylation,37,38 HSCs may have increased sensitivity to DNMT inhibitors in the event that the drug gains access into the DNA. Given that HSCs rely on DNMT1 for their survival, self-renewal, and repopulation potential,39,40 the mechanism of action of DNMT inhibitors on HSCs upon drug incorporation might also be a result of pharmacologic depletion of DNMT1 itself.
Because most HSCs are quiescent, we examined whether AZA itself will induce HSC proliferation. We observed that consecutive in vivo dosing of AZA resulted in pronounced depletion of mature cells and proliferation of HSCs and MPP cells. In vitro assays confirmed that AZA, at concentrations equivalent to plasma levels observed in patients, was directly toxic to both mouse and human proliferating HSCs. We thus hypothesized that AZA induces stress hematopoiesis, causing HSCs to leave their quiescent state, and in the setting of proliferating HSCs, the drug exerts direct cytotoxic effects. It follows that sequential doses of the hypomethylating agent, as is given clinically, is required to obtain HSC depletion. However, we recognize that the metabolites of AZA incorporate into both DNA and RNA,41,42 and there are likely additional AZA-mediated intra- and extracellular events that explain the activity of the drug on normal and clonally abnormal hematopoietic cells.
Having noted that AZA induces the death of HSPCs, and given that previous studies have shown that SCF can rescue hematopoiesis after irradiation-related injury,43-45 we hypothesized that the addition of a CD117 blocking antibody to AZA treatment might exacerbate the AZA-induced injury to HSCs. Indeed, our in vivo studies using ACK2-AZA showed increased apoptotic death of HSPCs, prolonged depletion of LT-HSCs, and robust engraftment of donor HSCs. Delivery of ACK2 in advance of AZA treatment was required to achieve these effects. Furthermore, we tested LT-HSC depletion with AZA plus a different anti-CD117 antibody, 2B8, which does not completely block hematopoiesis in vitro in the presence of SCF.31 The combination of ACK2 and 2B8 showed no enhanced efficacy for depleting LT-HSCs in vivo compared with AZA alone. Taken together, these findings support the important role of SCF-CD117 signal blockade in the synergy between ACK2 and AZA.
The potential clinical applications for this anti-CD117 antibody plus AZA combination are broad and span both nonmalignant and malignant hematologic disorders. On the basis of the mechanisms of action and our clinical experience, these 2 agents used as transplant conditioning are expected to have a substantially better safety profile compared with traditional alkylator or radiation-based regimens. Busulfan, the most commonly used alkylator in transplantations, has well-known acute and chronic toxicities, the latter of which are especially problematic for children. The acute risks of busulfan are pancytopenia, mucositis, seizure, and hepatic sinusoidal obstructive syndrome. Long-term sequelae include infertility, growth failure, pulmonary fibrosis, neurodevelopmental delays, and increased risk of cancer. By contrast, the major toxicities of AZA (myelosuppression and gastrointestinal symptoms such as nausea and vomiting) are acute. Long-term data on the effects of AZA in children are lacking, but the extensive experience with AZA in adults does not raise the same specter of irreversible tissue damage and mutagenesis as busulfan. With regard to anti-CD117 agents, our group recently reported on the safety and efficacy of the anti-CD117 antibody JSP191 used as single-agent conditioning before allogeneic HCT in children with SCID. All patients tolerated JSP191 well, without safety issues, and all patients showed evidence of LT-HSC engraftment.6,7 For other nonmalignant diseases (eg, sickle cell disease, thalassemia, Fanconi’s anemia), patients routinely present to transplantation with increased risk of transplant-related toxicities because of their inherent disease-specific comorbidities, including hepatic or renal dysfunction and iron overload.46 Importantly, achievement of mixed hematopoietic chimerism (on the order of >20%) is sufficient to provide curative benefit, particularly for hemoglobinopathies.47 A safer pretransplant conditioning regimen could permit more patients to qualify or electively pursue HCT as a curative option earlier in the disease course and significantly improve quality of life for long-term survivors of HCT.
The implications of our findings for hematologic malignancies include transplantation, but may extend to the use of ACK2-AZA as a therapeutic regimen. For myeloid malignancies, elimination of leukemic stem cells (LSCs) is essential for disease eradication.48 Despite the disease-modifying effects of AZA in patients with myeloid malignancies, single-agent AZA is insufficient for eliminating LSCs and therefore does not achieve durable remissions.49 By comparison, the combination of AZA with the B-cell lymphoma 2 (BCL-2) inhibitor venetoclax has proved to be more efficacious in AML by synergistic targeting of LSCs.50-52 We recently showed that the anti-CD117 mAb JSP191 depletes human MDS HSCs in MDS-xenografted mice.8 Future investigations will study the combination of anti-CD117 antibody and AZA on the eradication of MDS and AML.
In summary, our studies provide a platform for the expanded clinical use of AZA as a therapeutic agent, and for the use of a novel combination of AZA and anti-CD117 antibody in the setting of HCT, which may prove to be highly beneficial to the treatment of malignant and nonmalignant hematologic diseases in the near future.
Acknowledgments
The authors thank Jessica Poyser for excellent laboratory management, technical support of the studies, and animal husbandry; Steve Jurgens for technical assistance with flow cytometry and fluorescence-activated cell sorting; and Antonia M.S. Müller at the University Hospital in Zurich for critical reading of the manuscript and helpful discussions.
This work was supported by grants from the California Institute for Regenerative Medicine (DR2A-05365) (J.A.S.), the National Cancer Institute, National Institutes of Health (P01CA049605), and National Heart, Lung, and Blood Institute (OT2HL152830) (J.A.S.), the Gunn/Oliver Research Fund (J.A.S.), the D.K. Ludwig Fund for Cancer Research (J.A.S.), and the HL Snyder Medical Foundation (J.A.S.).
Authorship
Contribution: A.K.B. and W.W.P. conceived the project and designed the studies; A.K.B. performed experiments and analyzed the data; B.J.V. provided technical assistance; A.K.B. and J.A.S. wrote the manuscript; W.W.P., B.J.V., and J.R.L.-B. edited the manuscript; and J.A.S. supervised the project.
Conflict-of-interest disclosure: A.K.B., W.W.P., and J.A.S. are inventors on a patent that pairs anti-CD117 antibodies for HCT conditioning. J.A.S. is a cofounder, stockholder, and a director of Jasper Therapeutics, Inc. W.W.P. is a stockholder and employee of Jasper Therapeutics, Inc. The remaining authors declare no competing financial interests. Jasper Therapeutics, Inc has licensed a patent from Stanford University that includes the use of anti-CD117 antibodies in HCT conditioning.
Correspondence: Judith A. Shizuru, Stanford University School of Medicine, 269 Campus Drive West 2205a, Stanford CA, 94305; e-mail: jshizuru@stanford.edu.

