

Key Points
CD169+ MPs are proinflammatory in the myeloma microenvironment; deletion of MPs reduces dissemination of myeloma and increases survival.
IL-6 and TNFα mobilize myeloma from BM into the blood by increasing vascular leakiness and reducing adhesion by downregulating CD138.

Abstract
Multiple myeloma (MM) is a plasma cell malignancy characterized by the presence of multiple foci in the skeleton. These distinct tumor foci represent cycles of tumor growth and dissemination that seed new clusters and drive disease progression. By using an intratibial Vk*MYC murine myeloma model, we found that CD169+ radiation-resistant tissue-resident macrophages (MPs) were critical for early dissemination of myeloma and disease progression. Depletion of these MPs had no effect on tumor proliferation, but it did reduce egress of myeloma from bone marrow (BM) and its spread to other bones. Depletion of MPs as a single therapy and in combination with BM transplantation improved overall survival. Dissemination of myeloma was correlated with an increased inflammatory signature in BM MPs. It was also correlated with the production of interleukin-6 (IL-6) and tumor necrosis factor α (TNFα) by tumor-associated MPs. Exogenous intravenous IL-6 and TNFα can trigger myeloma intravasation in the BM by increasing vascular permeability in the BM and by enhancing the motility of myeloma cells by reducing the adhesion of CD138. Moreover, mice that lacked IL-6 had defects in disseminating myeloma similar to those in MP-depleted recipients. Mice that were deficient in TNFα or TNFα receptor (TNFR) had defects in disseminating MM, and engraftment was also impaired. These effects on dissemination of myeloma required production of cytokines in the radiation-resistant compartment that contained these radiation-resistant BM MPs. Taken together, we propose that egress of myeloma cells from BM is regulated by localized inflammation in foci, driven in part by CD169+ MPs.
Introduction
Multiple myeloma (MM) is a disseminated plasma cell (PC) malignancy in the bone marrow (BM) that leads to multiple bone lytic lesions, anemia, renal involvement, and immunodeficiency.1 Progression of MM2,3 and prognosis4,5 correlate with increased interleukin-6 (IL-6), a proliferation-inducing ligand (APRIL), and tumor necrosis factor α (TNFα). Indeed, some evidence suggests that inflammatory autoimmune diseases can promote disease progression.6-8 TNFα is associated with mononuclear cells and can expand myeloma in vitro9 and thus affect cell survival.10,11 In addition, TNFα enhances the migration of myeloma cells through the BM endothelial cell monolayer.12 However, blocking TNFα did not enhance survival because it had the unexpected consequence of increasing the level of TNFα, which muddled the interpretation of its role in MM.13 IL-6 can promote growth and survival factors for MM by decreasing apoptosis, mediating drug resistance,14,15 and promoting angiogenesis via VEGF in vitro.16
Dissemination of cancer cells throughout the skeletal system and extramedullary sites is a major cause of death in patients with MM. We recently showed that blockade of CD138 triggered the dissemination of myeloma, leading to disease progression.17 Extracellular vesicles released by MM cells also favor dissemination of cancer cells to distant bones.18
Materials and methods
Mice and treatments
C57BL/6 (B6) or CD45.1+ B6 mice were obtained from Charles River. IL-6−/− (#2650), TNFα−/− (#5540), and TNFα receptor (TNFR−/−) (#3243) strains were purchased from The Jackson Laboratory. CD169-diphtheria toxin receptor (CD169-DTR)21 mice were bred in-house. All mice were kept in specific pathogen-free animal facilities. The GFP-Vk14451 transplantable MM line was obtained by crossing Vk*MYC mice22 with mice that expressed EGFP under the control of a gamma1 promoter.23 Myeloma cells were inoculated intravenously (1 × 104) or intratibially (2 × 104).17 To deplete MPs, CD169-DTR mice were treated with 200 ng diphtheria toxin (DT) starting from day 0 (or day 14 in Figure 1C) twice per week for 2 weeks, except in Figure 5, where chimeric mice received DT on days 58 and 60 post tumor challenge. For chimeric animals, recipient mice were lethally irradiated with a single 950-rad dose and they received donor BM cells (1e6). After BM reconstitution (6-8 weeks), chimeric mice were intratibially inoculated with MM cells. The Institutional Animal Care and Use Committee of Albert Einstein College of Medicine approved all animal work (protocol 0000-1021).
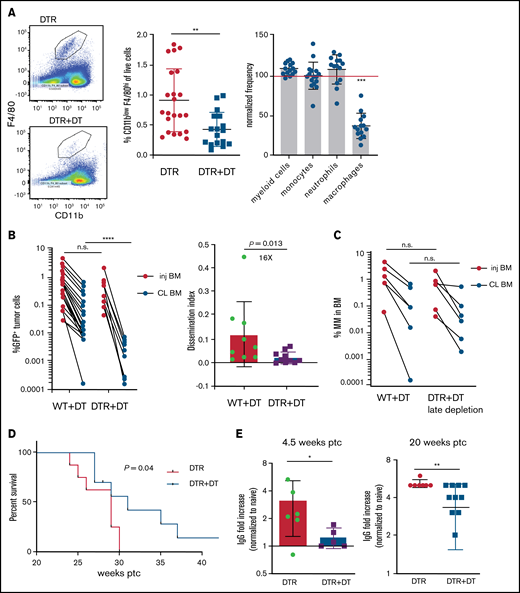
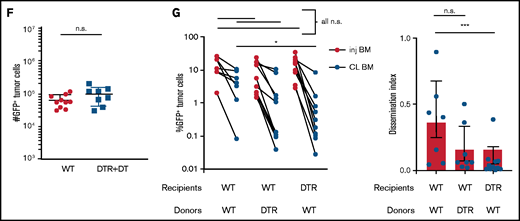
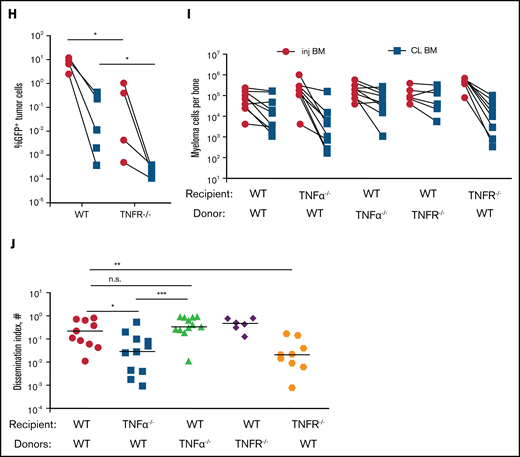
Tissue -resident MPs promote dissemination of myeloma and disease progression. (A) Sample plots and analysis of MP (CD11blowF4/80+) frequencies in the BM in CD169-DTR mice before and after treatment with DT; data were pooled from total myeloid cells (CD11b+), monocytes (CD11b+Ly6Chi), neutrophils (CD11b+Ly6Ghi), and MPs normalized to untreated controls. (B) Tumor burden analyzed in paired injected (inj; blue) and contralateral (CL; green) tibias of DT-treated CD169-DTR mice and control mice at 5 weeks after intratibial tumor inoculation and calculated dissemination index (ratio of myeloma burden in contralateral BM to that in injected BM). (C) Experiment setup as in panel B, with DT treatments starting at 2 weeks post tumor challenge (ptc). (D) Survival analysis (using Mantel-Cox test) of DT-treated or untreated CD169-DTR mice after intratibial tumor inoculation as in panel B. (E) Analysis of M-spike levels at 5 weeks and 20 weeks post tumor challenge in survival study shown in panel D. (F) Tumor burden was analyzed in DT-treated CD169-DTR (DTR+DT) mice and control (WT+DT) mice at 5 weeks after intravenous inoculation. (G) Analysis of tumor burden and dissemination index in chimeric hosts generated from lethal irradiation of recipient mice and reconstitution with donor BM cells, as labeled. Data from multiple experiments were pooled. All experiments were independently repeated 2 to 5 times, and each dot presents an individual mouse. Data comparisons were analyzed by using a Mann-Whitney t test. Error bars represent standard deviation. *P < .05; **P < .01; ***P < .001; ****P < .0001. n.s., not significant.
Tissue -resident MPs promote dissemination of myeloma and disease progression. (A) Sample plots and analysis of MP (CD11blowF4/80+) frequencies in the BM in CD169-DTR mice before and after treatment with DT; data were pooled from total myeloid cells (CD11b+), monocytes (CD11b+Ly6Chi), neutrophils (CD11b+Ly6Ghi), and MPs normalized to untreated controls. (B) Tumor burden analyzed in paired injected (inj; blue) and contralateral (CL; green) tibias of DT-treated CD169-DTR mice and control mice at 5 weeks after intratibial tumor inoculation and calculated dissemination index (ratio of myeloma burden in contralateral BM to that in injected BM). (C) Experiment setup as in panel B, with DT treatments starting at 2 weeks post tumor challenge (ptc). (D) Survival analysis (using Mantel-Cox test) of DT-treated or untreated CD169-DTR mice after intratibial tumor inoculation as in panel B. (E) Analysis of M-spike levels at 5 weeks and 20 weeks post tumor challenge in survival study shown in panel D. (F) Tumor burden was analyzed in DT-treated CD169-DTR (DTR+DT) mice and control (WT+DT) mice at 5 weeks after intravenous inoculation. (G) Analysis of tumor burden and dissemination index in chimeric hosts generated from lethal irradiation of recipient mice and reconstitution with donor BM cells, as labeled. Data from multiple experiments were pooled. All experiments were independently repeated 2 to 5 times, and each dot presents an individual mouse. Data comparisons were analyzed by using a Mann-Whitney t test. Error bars represent standard deviation. *P < .05; **P < .01; ***P < .001; ****P < .0001. n.s., not significant.
Antibodies and flow cytometry
The following antibodies were obtained from eBioscience, BioLegend, or BD Biosciences: anti-CD138 (281-2), anti-CD169 (SER-4), anti-CD11b (M1/70), anti-F4/80 (BM8), anti-Ly6G (1A8), and anti-CD206 (C068C2). For intracellular detection of cytokines, cells were incubated for 4 hours with brefeldin-A (BD Biosciences), stained for extracellular markers, and intracellularly stained with anti-TNFα (MP6-XT22) and anti-IL-6 (MP5-20F3).
Enzyme-linked immunosorbent assay
Paraprotein (IgG2a) was detected by sandwich enzyme-linked immunosorbent assay (ELISA); plates were coated with polyclonal anti-mouse immunoglobulin G (IgG) and anti-IgG2a-horseradish peroxidase (Jackson ImmunoResearch). The IgG2a fold increase was normalized to that of age-matched naïve mice.
Functional assays and intravital imaging
For intravasation measurements, tumor-bearing mice (4-6 weeks after tumor challenge) were bled (50 μL) before and at 2.5 or 24 hours after treatment with recombinant TNFα (rTNFα; 1 µg/100 μL phosphate-buffered saline [PBS; R&D Systems]) or IL-6 (64 ng/100 μL PBS; BioLegend). For experiments regarding vessel leakiness, mice were similarly treated with cytokines and given CD138-phycoerythrin (PE) intravenously 2 minutes before they were euthanized. Cells were then stained with CD138-BV450 ex vivo. Surgery and imaging with two-photon laser scanning microscopy (TPLSM) were described previously.17 For in vivo imaging experiments, mice were imaged before and after acute treatment with rTNFα (1 µg/100 µL PBS; R&D Systems) for 3.5 hours total. In vivo imaging experiments studied vascular leakage by intravenously administering Texas Red–conjugated dextran (70 kDa) followed by measuring parenchyma dextran uptake (pixels 25-50 microns from the bone surface).
RNA sequencing and analysis
Total RNA was extracted from samples by using RNeasy Plus Micro Kit (Life Technologies). Libraries were generated with SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing and Prep Kit (TakaraBio 634899). Libraries were sequenced on an Illumina HiSeq-4000 sequencer. The sequencing reads were aligned to the mouse genome (mm10/GRCm38) using the splice-aware STAR aligner,24 and counts were computed using the featureCounts program.25 Differentially expressed genes (DEGs) and principal component analysis (PCA) were computed using DESeq2 (version 1.26.0),26 based on twofold changes and multiple test adjusted P < .05. DEG overlaps among different MP subtypes were analyzed using InteractiVenn.27 Gene Ontology (GO) enrichment analysis was performed using ClueGO,28 with adjusted P < .05. Heatmaps were generated in RStudio (Ver1.1.383) using the pheatmap package (ver1.0.12). The RNA sequencing (RNA-seq) data have been deposited in the Gene Expression Omnibus database (accession number GSE176385).
Ligand-receptor interaction analysis
Potential ligand-receptor interactions29 were used to identify ligands on the basis of upregulated DEGs in 3 MP subsets (naive MPs, 2 subsets of tumor associated macrophages [TAMs] either in contact with myeloma [TAM IC] or TAMs not in contact [TAM NIC]) and to determine whether cognate receptors were significantly upregulated in myeloma cells vs control PCs. The interaction strength for each identified ligand-receptor pair was computed by multiplying the average expression of a ligand in the MP subsets with the average expression of the receptor in the myeloma samples. In a similar manner, computed ligands that were upregulated in myeloma samples were compared with control PCs and their corresponding receptors that were significantly upregulated in the TAM IC and TAM NIC subsets vs naïve MPs. The resulting ligand-receptor pairs were clustered by their interaction scores and were analyzed for enriched GO-terms using Toppgene.30
Results
Tissue-resident MPs promote dissemination of myeloma and disease progression
To address the role of TAMs in the context of growth and dissemination of MM in vivo, we used a green fluorescent protein (GFP)–expressing Vk*MYC murine myeloma model that recapitulates the biological and clinical features of human MM and develops in the BM of immunocompetent C57BL/6 mice.17,22 In mice expressing the CD169-DTR allele,21 systemic administration of DT specifically ablates CD169+ tissue-resident MPs, but BM monocytes or neutrophils are not affected (Figure 1A).
To explore the functional role of CD169+ MPs in MM, we inoculated CD169-DTR mice with GFP-expressing Vk*MYC murine myeloma cells via intratibial injection. Previously, we found that intratibial injection restricted the growth of myeloma cells to the injected tibia at early stages; myeloma cells then spread to other sites and that allowed us to monitor growth (in a primary injected tibia) and dissemination (to the contralateral tibia).17 Depletion of CD169+ MPs did not affect tumor burden in the BM of the injected tibia, but there was a marked reduction in tumor size in the contralateral BM, which had dissemination 16-fold lower compared with controls (Figure 1B). However, if administration of DT was delayed until 2 weeks post tumor challenge, depletion of MPs was not effective, suggesting that CD169+ macrophages play a role in early spreading of myeloma (Figure 1C). Early depletion of CD169+ MPs extended survival (from 29 to 31 days; P = .04) and reduced the hazard ratio (from 1 to 0.42; 95% confidence interval, 0.14-1.2) compared with control mice (Figure 1D). Moreover, MP-depleted mice maintained significantly lower M-spike compared with that in untreated animals, which indicates that overall tumor burden was reduced (Figure 1E).
Spreading of myeloma is a multistep process that requires both egress from the BM and subsequent re-engraftment. To understand which steps were dependent on MPs, CD169-DTR mice were intravenously inoculated with myeloma cells bypassing egress from the BM. Depletion of MPs had no effect on myeloma burden when myeloma cells were intravenously transferred, suggesting that egress from the BM is a critical step regulated by MPs (Figure 1F) and that BM re-engraftment is independent of MPs.
CD169+ BM MPs have previously been shown to be tissue resident and radiation resistant.31-33 To determine whether TAMs were also tissue-resident MPs, we reconstituted lethally irradiated wild-type (WT) recipients with CD169-DTR BM (DTR→WT mice), or generated the reverse chimeric mice (WT→DTR mice). After reconstitution, mice were inoculated intratibially with myeloma cells and treated with DT to deplete CD169+ MPs. Depletion of tissue-resident MPs reduced the number of CD169+ MPs in the BM but not the total number of MPs in the BM (supplemental Figure 1A-B). However, only depletion of these radiation-resistant, tissue-resident MPs (WT→DTR mice) had significant defects in the process of spreading myeloma to other bones as found in intact CD169-DTR mice (Figure 1G); this was in contrast to control mice (WT→WT) or depletion of hematopoietic-derived CD169+ cells (DTR→WT). These results confirm that depletion of MPs using the CD169-DTR model targets a unique tissue-resident MP population31-33 that is critical for regulating dissemination of myeloma.
TAMs exhibit proinflammatory phenotype in BM MM cluster
By using TPLSM in the tibial BM of mice, we found that GFP+ Vk*MYC myeloma cells grew as clustered foci the same way it did in patients.17 At early stages, myeloma tumors grew as clusters, but surprisingly, tumors cells were not attached to one another. Rather, they were in close contact with CD169+ MPs (Figure 2A; supplemental Movie 1). We also observed long GFP+ protrusions (similar to tunneling nanotubes34 ) connecting myeloma cells that were present in advanced tumors (>10% of BM). These nanotubes were dynamic and could be seen (by using TPLSM) transferring GFP+ material from myeloma cells to neighboring autofluorescent MPs (Figure 2B; supplemental Movie 2). On the basis of the imaging, we posited that TAMs in close contact with myeloma cells may become GFP+ through uptake or transfer of myeloma cytoplasm by various means, and they were detectable as a GFPlow MP population by flow cytometry (Figure 2C). Further characterization of GFPlow TAMs revealed a proinflammatory phenotype based on CD206low expression.35 GFPlowF4/80+ MPs had significantly lower CD206 expression compared with GFP– MPs within the same tibia (Figure 2C), and as tumor burden increased, more MPs shifted to the proinflammatory phenotype (Figure 2D). To verify that TAMs were indeed proinflammatory, we measured the intracellular production of the TNFα and IL-6 inflammatory type 1 cytokines. Indeed, cytokine levels were elevated in TAMs compared with naïve control BM MPs (Figure 2E), and TNFα production was correlated with the CD206 subset (supplemental Figure 1C). Although TNFα-producing MPs increased with the growth of myeloma (Figure 2F), additional (CD11b+) myeloid cells became major producers as tumors continued to grow (supplemental Figure 1D). Concurrently, MP numbers decreased as myeloma increased, which may also contribute to the decreased role of MPs in the tumor microenvironment (supplemental Figure 1E). Taken together, these data suggest that BM TAMs have an inflammatory phenotype in myeloma foci.
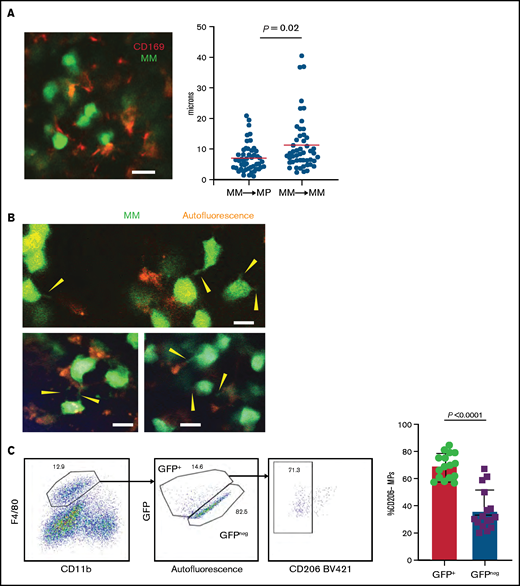
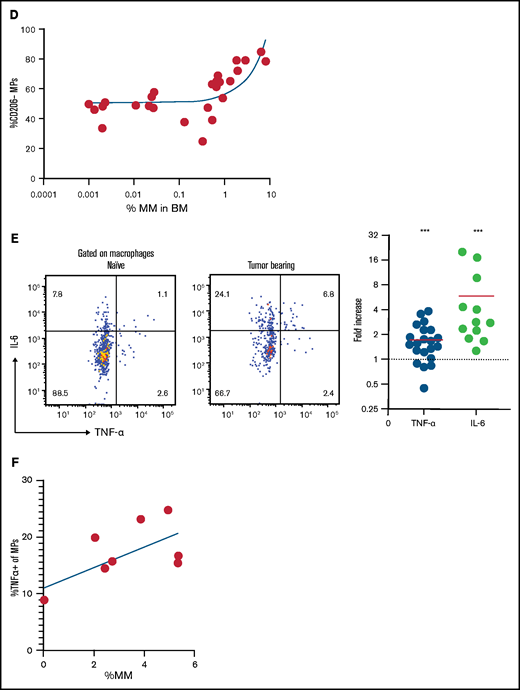
TAMs exhibit proinflammatory phenotype in BM cluster of MM cells. (A-B) Intravital live imaging of GFP+ myeloma cells (green) in the injected tibia at 2 weeks after inoculation (n = 2 mice). (A) 3D analysis of distances between myeloma cells and anti-CD169-PE–labeled MPs (red) within a small cluster. (B) Examples of myeloma nanotube structures extending to BM autofluorescent (MP) cells (red). Yellow arrows show transfer of GFP signals from MM cells to surrounded microenvironment cells. (C) Gating strategy to identify GFP+ and GFP– BM MPs and the CD206 subset and analysis of CD206– (M1-like) subset of GFP+ and GFP– BM MPs in tumor-bearing mice. (D) Kinetics of CD206– MP subset as a function of tumor burden in the BM. (E) Samples of frequency of intracellular IL-6 and TNFα production in MPs, with fold increase in tumor-bearing mice normalized to that of naïve mice, analyzed by a Wilcoxon test. (F) Plot of TNFα-producing BM MPs vs tumor burden in the BM. All experiments were independently repeated at least 2 times; data were pooled and comparisons were analyzed by using a Mann-Whitney t test. Scale bars, 20 μm. *** is P < .001. Errors bars in panel C represent standard deviation. Horizontal lines in panels A and E are mean values.
TAMs exhibit proinflammatory phenotype in BM cluster of MM cells. (A-B) Intravital live imaging of GFP+ myeloma cells (green) in the injected tibia at 2 weeks after inoculation (n = 2 mice). (A) 3D analysis of distances between myeloma cells and anti-CD169-PE–labeled MPs (red) within a small cluster. (B) Examples of myeloma nanotube structures extending to BM autofluorescent (MP) cells (red). Yellow arrows show transfer of GFP signals from MM cells to surrounded microenvironment cells. (C) Gating strategy to identify GFP+ and GFP– BM MPs and the CD206 subset and analysis of CD206– (M1-like) subset of GFP+ and GFP– BM MPs in tumor-bearing mice. (D) Kinetics of CD206– MP subset as a function of tumor burden in the BM. (E) Samples of frequency of intracellular IL-6 and TNFα production in MPs, with fold increase in tumor-bearing mice normalized to that of naïve mice, analyzed by a Wilcoxon test. (F) Plot of TNFα-producing BM MPs vs tumor burden in the BM. All experiments were independently repeated at least 2 times; data were pooled and comparisons were analyzed by using a Mann-Whitney t test. Scale bars, 20 μm. *** is P < .001. Errors bars in panel C represent standard deviation. Horizontal lines in panels A and E are mean values.
Proinflammatory TNFα and IL-6 cytokines promote dissemination of MM
We next tested whether these cytokines could mobilize myeloma directly. Within 2 hours after intravenous treatment with cytokines, myeloma cells were rapidly mobilized into the blood (Figure 3A). By 24 hours after treatment, mobilization of MM increased to fivefold above that with treatment using PBS (Figure 3B), possibly through amplification via autocrine or paracrine signaling. At 2 weeks after intratibial injection of myeloma cells, we know that the majority of the myeloma cells are in the injected tibia; therefore, mobilization is likely occurring from that site.17 To confirm that treatment was acting on the BM, we imaged myeloma cells by intravital TPLSM before and after treatment with cytokines and could see increased cell motility (Figure 3C) and rapid egress after treatment (supplemental Movie 3).
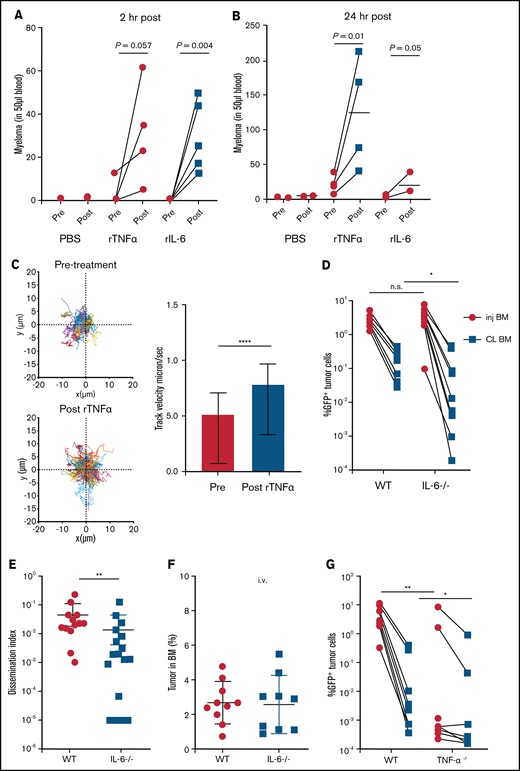
Proinflammatory cytokines TNFα and IL-6 promote dissemination of MM. (A) Analysis of circulating tumor cells in tumor-bearing mice (4-6 weeks post tumor challenge), before (pre) and 2 hours (hr) after (post) treatment for 2 days with recombinant cytokines rTNFα or rIL-6. (B) Analysis of circulating tumor cells in tumor-bearing mice (4-6 weeks post tumor challenge) at 24 hours after treatment for 2 days with rTNFα or rIL-6. (C) Cell tracks of myeloma cells (n = 99) after time-lapse imaging in the BM focus before and 0 to 2 hours after treatment with rTNFα (1 µg) and analysis of their track speeds. (D-E) Analysis of tumor burdens in tibias and dissemination index in IL-6 and WT hosts after intratibial tumor inoculation. (F) Analysis of tumor burden in bones after intravenous (i.v.) tumor inoculation. (G-H) Comparison of tumor burdens (as in panel D) in TNFα−/−, TNFR−/−, and WT control mice. (I-J) Analysis of tumor burden and dissemination index in chimeric hosts, generated from lethal irradiation of recipient mice and reconstitution with donor BM cells as labeled. All experiments were independently repeated at least 2 times. Data points represent individual mice, and data from multiple experiments were pooled; comparisons were analyzed by using a Mann-Whitney t test. *P < .05; **P < .01; ***P < .001; ****P < .0001. Thick horizontal lines are means throughout; error bars are standard deviation in panels C and F and SEM in panel E.
Proinflammatory cytokines TNFα and IL-6 promote dissemination of MM. (A) Analysis of circulating tumor cells in tumor-bearing mice (4-6 weeks post tumor challenge), before (pre) and 2 hours (hr) after (post) treatment for 2 days with recombinant cytokines rTNFα or rIL-6. (B) Analysis of circulating tumor cells in tumor-bearing mice (4-6 weeks post tumor challenge) at 24 hours after treatment for 2 days with rTNFα or rIL-6. (C) Cell tracks of myeloma cells (n = 99) after time-lapse imaging in the BM focus before and 0 to 2 hours after treatment with rTNFα (1 µg) and analysis of their track speeds. (D-E) Analysis of tumor burdens in tibias and dissemination index in IL-6 and WT hosts after intratibial tumor inoculation. (F) Analysis of tumor burden in bones after intravenous (i.v.) tumor inoculation. (G-H) Comparison of tumor burdens (as in panel D) in TNFα−/−, TNFR−/−, and WT control mice. (I-J) Analysis of tumor burden and dissemination index in chimeric hosts, generated from lethal irradiation of recipient mice and reconstitution with donor BM cells as labeled. All experiments were independently repeated at least 2 times. Data points represent individual mice, and data from multiple experiments were pooled; comparisons were analyzed by using a Mann-Whitney t test. *P < .05; **P < .01; ***P < .001; ****P < .0001. Thick horizontal lines are means throughout; error bars are standard deviation in panels C and F and SEM in panel E.
To confirm these results, IL-6−/− mice were intratibially injected with myeloma cells and tumor burden was analyzed . Although the size of the myeloma tumor in the injected tibia was similar, dissemination of myeloma to the contralateral tibia was significant reduced (Figure 3D-E). Importantly, when myeloma cells were intravenously administered, tumor burden in the bones of IL-6−/− recipients was similar to that in WT hosts (Figure 3F), indicating that egress from the BM was dependent on IL-6. These results mirrored the pattern of the growth and dissemination of myeloma seen after MP depletion (Figure 1B,F).
Next, we analyzed the role of TNFα in the growth and spreading of myeloma by intratibially inoculating tumors into TNFα−/− and WT mice. Strikingly, there were severe defects in growth or engraftment of myeloma cells in TNFα−/− mice (Figure 3G), suggesting a major role for TNFα in the development of MM. To determine whether TNFα was signaling in myeloma cells or acting indirectly on the BM microenvironment cells, we transferred myeloma by intratibial injection into mice deficient for TNFR1 and TNFR2 (TNFR−/−) or control mice. Growth and engraftment of myeloma were abrogated in the TNFR−/− recipients (Figure 3H), similar to the results in TNFα−/− mice, suggesting that TNFα acts indirectly on non-tumor BM cells to facilitate growth and engraftment of myeloma.
Although IL-6 and TNFα are widely produced, the highest expression is restricted to hematopoietic compartment subsets such as myeloid cells, natural killer cells, and B cells. However, because we found that the radiation-resistant compartment promotes dissemination of myeloma and this seemed to be functionally similar to TNFα and IL-6 deficiencies, we tested whether production of TNFα by radiation-resistant cells was required for myeloma to spread to other bones. To accomplish this, we generated BM chimeric mice that lacked production of either cytokine in the radiation-resistant compartment or in the donor-derived compartment. As controls for these chimeras, we irradiated and reconstituted WT BM into WT mice. After reconstitution of the BM, mice were intratibially injected with myeloma, and tumor growth was assessed 6 weeks after tumor challenge. In TNFα−/− recipients reconstituted with WT BM (WT→TNFα−/−), the dissemination index and tumor size in the contralateral BM was significantly lower compared with that in control groups (Figure 3I-J), indicating that spreading of myeloma required TNFα production in the radiation-resident compartment, which includes but is not limited to the CD169+ BM MPs.
Although we found that TNFα acts indirectly on the host to control engraftment of myeloma, we generated TNFR chimeras to determine which BM compartment was an important TNFR-carrying compartment in the spread of MM. WT or TNFR−/− donors received TNFR−/− or WT BM, respectively (TNFR−/−→WT; WT→TNFR−/−) and were challenged with MM after reconstitution. Tumor growth in contralateral BM and dissemination index were reduced in the WT→TNFR−/− group compared with controls, WT→WT, and TNFR−/−→WT groups (Figure 3I-J). These data indicate that TNFα produced by the radiation-resistant compartment of BM promotes dissemination of MM. We also noted that myeloma spreading to the contralateral tibia, even in WT→WT hosts, was considerably more advanced compared with that in nonirradiated WT hosts (Figure 1B), suggesting that radiation damage increases spreading. Altogether, these data suggest that TNFα-producing radiation-resistant BM cells, which include tissue-resident MPs, promote dissemination of MM.
In BM chimeras that lacked TNFα or TNFR in the radiation-resistant compartment, IL-6–producing MPs were decreased (supplemental Figure 2A), suggesting that there was synergy between IL-6 and TNFα signaling. Ex vivo treatment of BM MPs with rTNFα from naïve mice stimulated production of cytokines IL-6 or TNFα or both. However, this process was increased when compared with BM MPs from myeloma-bearing mice (supplemental Figure 2B), suggesting that these pathways may interact.
Increased vascular leakage and reduced surface CD138 contribute to inflammation-enhanced dissemination
Because TNFR expression was required in radiation-resistant cells to promote dissemination of myeloma, we hypothesized that inflammation may be increasing vascular permeability. To test this, we used TPLSM to measure leakage of intravenously administered Texas Red-labeled dextran in the BM microenvironment of tumor-bearing mice. Dextran leakage and uptake by parenchymal phagocytes was higher within myeloma clusters compared with surrounding regions, which indicates a local effect (Figure 4A).
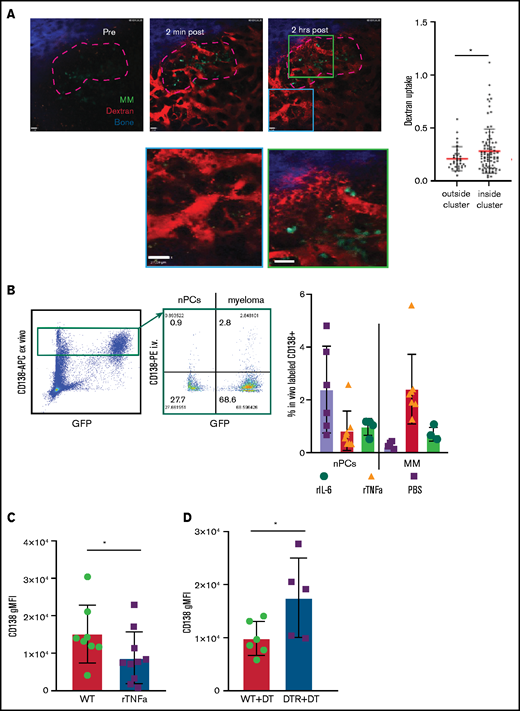
Increased vascular leakage and reduced surface CD138 contribute to inflammation-enhanced dissemination. (A) Snapshots from intravital time-lapse images taken before, 2 minutes after, and 2 hours after intravenous administration of Texas Red dextran in the myeloma focus (yellow dashed outline) in the tibia. Insets show dextran leakage in regions within the focus (green) and outside the focus (cyan). (B) Flow cytometric analysis of anti-CD138-PE in vivo labeling of polyclonal normal PCs (nPCs) and myeloma cells (MM) in the tibia BM, with ex vivo co-staining with anti-CD138-APC. (C) Flow cytometric analysis of surface expression of CD138 on myeloma cells with and without in vitro treatment with rTNFα. (D) Comparison of surface expression of CD138 on myeloma cells in the BM of DT-treated or untreated CD169-DTR recipients. Scale bars represent 22 μm. All error bars are standard deviation, with horizontal bars reflecting the mean. *P < .05. APC, allophycocyanin; gMFI, geometric mean fluorescence intensity.
Increased vascular leakage and reduced surface CD138 contribute to inflammation-enhanced dissemination. (A) Snapshots from intravital time-lapse images taken before, 2 minutes after, and 2 hours after intravenous administration of Texas Red dextran in the myeloma focus (yellow dashed outline) in the tibia. Insets show dextran leakage in regions within the focus (green) and outside the focus (cyan). (B) Flow cytometric analysis of anti-CD138-PE in vivo labeling of polyclonal normal PCs (nPCs) and myeloma cells (MM) in the tibia BM, with ex vivo co-staining with anti-CD138-APC. (C) Flow cytometric analysis of surface expression of CD138 on myeloma cells with and without in vitro treatment with rTNFα. (D) Comparison of surface expression of CD138 on myeloma cells in the BM of DT-treated or untreated CD169-DTR recipients. Scale bars represent 22 μm. All error bars are standard deviation, with horizontal bars reflecting the mean. *P < .05. APC, allophycocyanin; gMFI, geometric mean fluorescence intensity.
To confirm these results, tumor-bearing mice were treated with either rTNFα or IL-6 and, the next day, they were IV injected with anti-CD138-PE and were euthanized 2 minutes later. This approach will selectively label only vascular cells, but we reasoned that if cytokines were enhancing vascular leakiness in myeloma clusters, we would see increased labeling of myeloma cells only, because PCs are diffusely distributed throughout the BM. Treatment with both recombinant IL-6 and TNFα increased frequencies of PE+ cells compared with those in untreated tumor-bearing mice (Figure 4B). Moreover, TNFα increased labeling of MM cells and not PCs, suggesting that inflammation-induced permeability was spatially restricted to myeloma clusters.
Previously, we showed that expression of CD138 promotes retention of myeloma cells in the BM, and blocking CD138 rapidly mobilizes myeloma17 ; thus, we tested whether TNFα contributes to an increase in the motility and mobilization of myeloma through downregulation of CD138. Indeed, treatment with recombinant TNFα ex vivo reduced the surface expression of CD138 by 50% compared with that in controls (Figure 4C) gated on live cells (supplemental Figure 2C). In addition, depletion of CD169+ MPs (as in Figure 1), increased the expression of CD138 on myeloma cells in vivo in the BM compared with myeloma cells in control mice (Figure 4D), suggesting that TAMs may regulate retention of myeloma via CD138. Taken together, these data suggest that proinflammatory cytokines IL-6 and TNFα promote dissemination of tumors by both directed and indirect pathways.
Tissue-resident MPs contribute to MM relapse after irradiation therapy
Relapse in patients with MM after current treatments that include BM irradiation remains a significant challenge. We hypothesized that tissue-resident radiation-resistant MPs might be involved in recurrence of MM and increased spreading after irradiation and BM transplantation (Figure 3I-J). We assessed the effects of MP depletion after MM relapse after irradiation (Figure 5A). We compared the level of M-spike at week 8 after irradiation and found decreased tumor burden in MP-depleted mice compared with control relapsed mice (Figure 5B). In addition, survival rate was significantly improved in the group (Figure 5C), suggesting that tissue-resident MPs were also involved in promoting MM relapse.
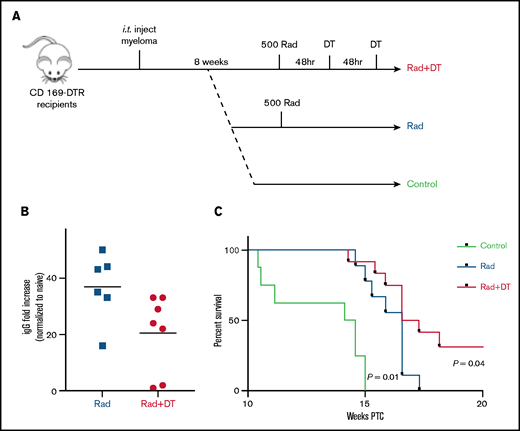
Tissue-resident MPs contribute to MM relapse after irradiation therapy. (A) Experimental design and groups . (B) Analysis of systemic tumor burden based on serum M-spike 8 weeks after irradiation normalized to serum from naïve mice. Bars represent mean. (C) Survival curve calculated by using the log-rank test (n = 8-9 mice per group).
Tissue-resident MPs contribute to MM relapse after irradiation therapy. (A) Experimental design and groups . (B) Analysis of systemic tumor burden based on serum M-spike 8 weeks after irradiation normalized to serum from naïve mice. Bars represent mean. (C) Survival curve calculated by using the log-rank test (n = 8-9 mice per group).
TAMs have unique gene expression signatures
Finally, to extend our analysis of TAM programming, we analyzed TAM transcriptomes by RNA-seq. By using fluorescence-activated cell sorting (FACS), we purified TAMs from tumor-bearing mice that were in contact (TAM-IC) or not in contact (TAM-NIC) with myeloma on the basis of GFP expression (supplemental Figure 2D) as well as BM MPs from naïve mice as controls for comparison. In addition, Vk*MYC myeloma cells and polyclonal (CD138hiB220low) BM PCs from naïve mice were also sorted by FACS and were analyzed to investigate myeloma-MP interactions. Sorted MPs were clustered into distinct groups based on PCA (Figure 6A). Next, we performed pairwise comparisons of the numbers of DEGs shared and uniquely expressed by the 3 MP subsets and found that a majority of DEGs were specific to each subset, even when comparing the 2 different TAM subsets (Figure 6B). To better understand this distinct type of expression, we clustered the 3 paired-wise DEGs into 5 clusters based on their expression among the 3 MP groups (Figure 6C; supplemental Tables 1-3). GO enrichment analysis was used to identify biological processes upregulated in the 2 TAM subsets (vs naïve MPs) (Figure 6D). We found that both TAM subsets had upregulated inflammatory and cytokine pathways, including TNFα, IL-6, and interferon γ (IFNγ) response, activated chemotaxis and migratory pathways, and elevated general immune activation. In addition, genes that are related to blood vessel development were highly enriched, which may help modulate vessel permeability (Figure 4). T-cell activation genes were enriched in TAM-NIC. Conversely, for genes upregulated only in TAM-IC, tube development and MP activation were among the processes most enriched. The TAM-IC subset also had various metabolic and transporter pathways that were activated, suggesting homeostatic changes in myeloma foci.
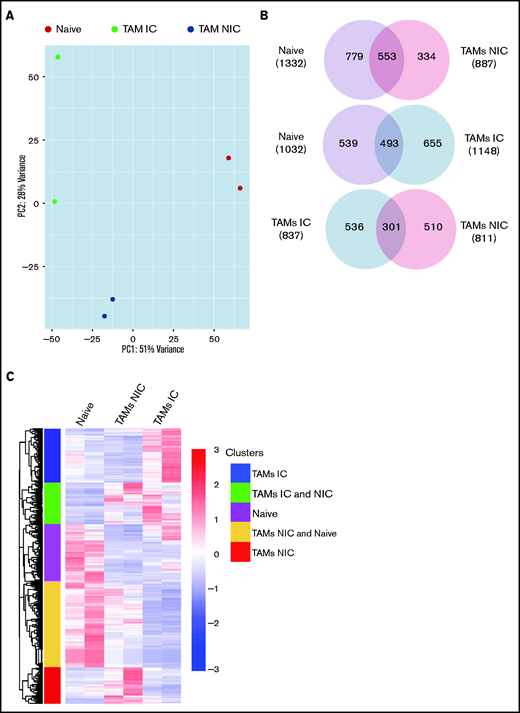
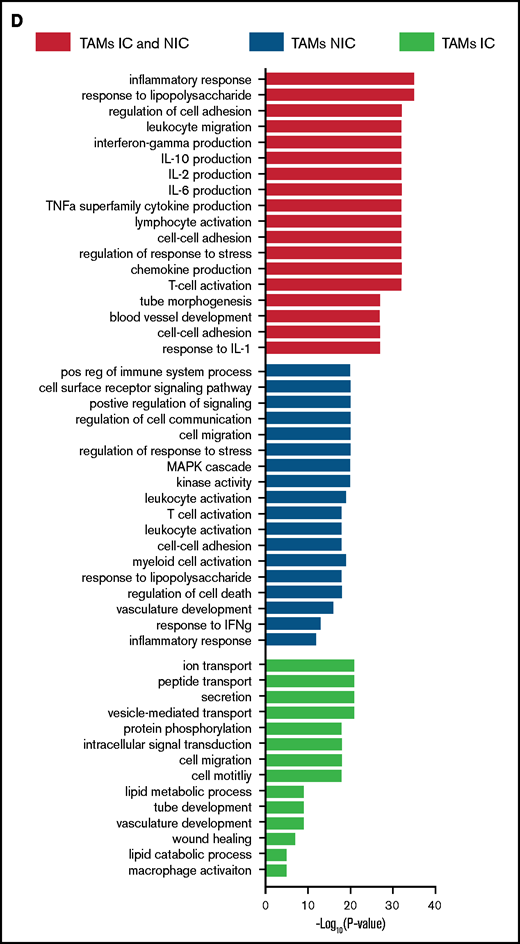
TAMs have unique gene expression signatures. (A) Principal component analysis plot comparing transcriptomes of naïve MPs, TAM-ICs, and TAM-NICs. (B) Venn diagrams comparing DEGs between 3 MP subsets. (C) Unsupervised clustering of all DEGs from 3 pair-wise comparisons: TAM IC vs TAM NIC, TAM IC vs MP, and TAM NIC vs MP. (D) GO analysis (biological process terms) for gene clusters upregulated commonly or uniquely in the 2 TAM subsets vs MPs.
TAMs have unique gene expression signatures. (A) Principal component analysis plot comparing transcriptomes of naïve MPs, TAM-ICs, and TAM-NICs. (B) Venn diagrams comparing DEGs between 3 MP subsets. (C) Unsupervised clustering of all DEGs from 3 pair-wise comparisons: TAM IC vs TAM NIC, TAM IC vs MP, and TAM NIC vs MP. (D) GO analysis (biological process terms) for gene clusters upregulated commonly or uniquely in the 2 TAM subsets vs MPs.
TAMs and MM have unique cell-cell interactions
On the basis of the MM-MP contacts that we observed and the changes in MP transcriptomes, we next analyzed the expression of cognate receptor-ligand pairs on both MPs and MM to reveal signaling that may underly TAM reprogramming and reveal MP ligands potentially interacting with MM cells. We determined the upregulated surface receptors on MM cells compared with PCs and cognate ligands on MP subsets to compute an interaction score for each ligand-receptor pair (Figure 7A; supplemental Table 4). This analysis found that MM:TAM-IC had the most putative interactions; multiple integrin interactions were detected, including ADAM-family metalloproteases, and FGF-family proteins. For the MM:TAM-NIC cluster, we found additional integrin interactions and soluble factors such as VEGFA, which could play a role in vascular remodeling. For both TAM subsets, additional integrin ligands and WNT cognate interactions were found, which are known to promote the development of MM.36 GO-term analysis of the receptor-ligand interactions among TAM subsets found that cell motility adhesion processes were broadly represented in the MM:TAM clusters; however, MM:TAM-IC had higher P values and involved more genes in these terms (Figure 7B).

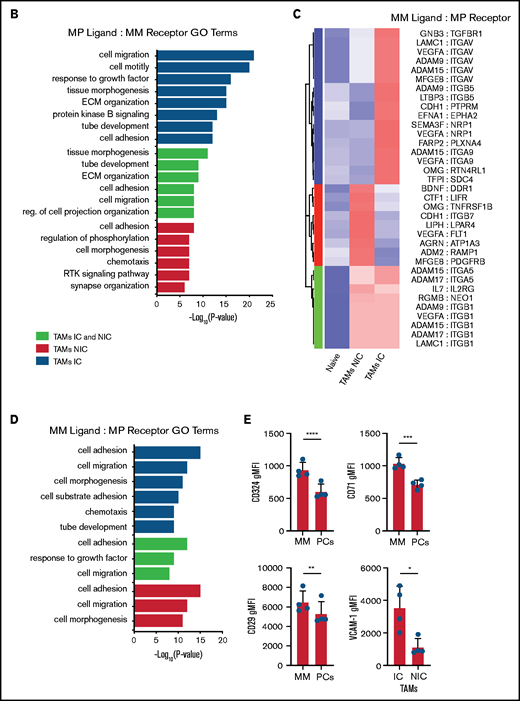
TAMs and MM have unique cell-cell interactions . (A) Upregulated MP ligand-MM receptor pairs clustered by interaction scores. (B) GO-term analysis of the 3 clusters of ligand-receptor pairs in panel A. (C) Upregulated MM ligand-MP receptor pairs clustered by interaction scores. (D) GO-term analysis of the 3 clusters of ligand-receptor pairs in panel C. (E) Flow cytometric analysis of surface expression of CD324, CD71, and CD29 on MM cells (CD138hiGFPhi) vs PCs (CD138hiB220–GFP–), and VCAM-1 expression on TAM-IC vs TAM-NIC cells using Mann-Whitney t tests (n = 3 mice) with similar results from a second experiment (not shown). *P < .05; **P < .01; ***P < .001; ****P < .0001. Error bars represent standard deviation.
TAMs and MM have unique cell-cell interactions . (A) Upregulated MP ligand-MM receptor pairs clustered by interaction scores. (B) GO-term analysis of the 3 clusters of ligand-receptor pairs in panel A. (C) Upregulated MM ligand-MP receptor pairs clustered by interaction scores. (D) GO-term analysis of the 3 clusters of ligand-receptor pairs in panel C. (E) Flow cytometric analysis of surface expression of CD324, CD71, and CD29 on MM cells (CD138hiGFPhi) vs PCs (CD138hiB220–GFP–), and VCAM-1 expression on TAM-IC vs TAM-NIC cells using Mann-Whitney t tests (n = 3 mice) with similar results from a second experiment (not shown). *P < .05; **P < .01; ***P < .001; ****P < .0001. Error bars represent standard deviation.
We hypothesized that the MM microenvironment may be contributing to TAM reprogramming, although these factors are unknown. We repeated the procedure to identify which MM ligands may be acting on TAM receptors (Figure 7C; supplemental Table 5). MM cells produced numerous integrin ligands whose receptors were highly expressed in TAMs, particularly in TAM-ICs. In addition, multiple neuropilin1 ligands (VEGFA and SEMA3F) and their receptors were highly enriched. Within the TAM-NIC and in shared TAM pathways, we found additional cytokines, VEGFA pathways that could promote blood vessel reorganization, and ADAM metalloproteases that could reorganize the extracellular matrix and promote egress (Figure 7D). We validated that the ligand-receptor pair VCAM-1:VLA-4 was upregulated on TAMs and MM cells as well as some other MM surface receptors (CD71, CD324) (Figure 7E). Targeting some of these factors such as VEGFA,37 FGF2,38 and TFRC39 has already been shown to provide some effect in solid tumors. Because these cognate interactions are hypothetical, their in vivo function remains to be determined.
Discussion
Our study provides novel insights into the functional role of TAMs in the BM during the growth and progression of myeloma. In particular, our model demonstrates that TAMs play a role in dissemination rather than growth, which is linked with worse survival, thus highlighting their importance in disease. A recent study showed the functional importance of MPs in myeloma progression.40 By using a clodronate liposome treatment, which ablated a wide range of phagocytes and MPs and also induced high levels of systemic inflammation, Opperman et al40 found reduced growth and engraftment of myeloma by using intravenous administration of the 5TGM1 MM model. In contrast, by using the CD169-DTR depletion system, which triggers a noninflammatory apoptotic cell death only in CD169+ MPs, we found that radiation-resistant cells are specifically required for dissemination of myeloma. Our results are complementary to those of the previous study, but they also identify inflammatory cytokines as the putative mechanism by which MPs promote progression of myeloma.
Although proinflammatory cytokines such as TNFα, IL-6, and IL-1 have long been known to be elevated in MM patients,41 it is believed their primary function is directly on myeloma cells to promote their survival. Here we identify inflammatory cytokines as key inducers of myeloma egress. The notion that BM TAMs trigger dissemination of myeloma is novel and possibly specific to blood cancers like myeloma. TAMs in breast cancer42 have increased invasion through CSF-VEGF signaling, but we found that BM MPs promote dissemination most likely through the production of TNFα and IL-6 by at least 2 mechanisms. First, TNFα promotes dissemination of tumors by directly triggering CD138 downregulation on myeloma cells. We previously found that CD138 promotes retention in the BM, and by blocking CD138, myeloma cells could be mobilized, which leads to their dissemination to other bones and thus larger overall tumor burden. Second, we found that rTNFα increased vascular permeability in the BM, and this effect was particularly localized in myeloma clusters. This suggests that these TAMs and tumor-associated endothelium are primed for leakiness. Increased vascular permeability in BM has been reported in other blood cancers but the molecular mechanisms were not defined.43 This also points to a new role for IL-6 in promoting dissemination of myeloma. Although IL-6 has been shown to promote the survival of myeloma in certain models, we have found that survival and growth of Vk*MYC cells is independent of IL-6, likely because of the overexpression of MYC in a STAT3-independent manner,17 which allows us to identify this additional role for IL-6. The requirements of TNFα expression and TNFR signaling in the host for engraftment of myeloma suggest additional cell-extrinsic roles for TNFα in myeloma seeding. We propose that radiation-resistant MPs are contributing to local inflammation, but we cannot rule out the potential for other radiation-resistant cells to produce TNFα and IL-6. Indeed, as tumors increased in size, radiation-sensitive myeloid cells were also producing high levels of TNFα.
One clear limitation of our study is that it was conducted in mice using only the Vk*MYC model, and it remains to be seen how this translates to patients. However, a recent study by de Jong et al44 focused on the role of inflammation in the myeloma microenvironment and potential roles for TNFα and many receptor-ligand pairs. This inflammatory signature remains even after induction therapy. de Jong et al suggested that T cells could be potential mediators of these proinflammatory cytokines, but they did not characterize BM MPs in their study. Thus, it remains to be seen whether MPs play the same sort of key role in patients as they do in mice. There is likely a redundancy in the inflammatory cells acting in the BM, particularly as tumors become more advanced, but all of them may have similar effects on the dissemination and spreading of tumors.
Overall, this study highlights the overlooked importance of how the spreading of myeloma cells is linked with disease progression. Interestingly, we find that dissemination is faster in chimeric recipient mice compared with nonchimeric recipient mice, which may be the result of damage and reprogramming of the BM microenvironment and vasculature after radiation. In these relapse settings, in which therapies have already damaged the BM microenvironment, targeting dissemination, as we did with MP depletion, may be a critical last defense against rapid tumor progression. Indeed, radiation may be promoting proinflammatory MPs45 and proinflammatory monocytes,46 thus accelerating the rate of relapse by promoting dissemination. We can devise better treatments by understanding how they regulate not only tumor burden but also tumor mobilization.
Acknowledgments
This work was funded in part by grants from the National Heart, Lung, and Blood Institute, National Institutes of Health (RO1 HL141491) (D.F.) and the National Cancer Institute, Albert Einstein Cancer Center core facilities (P30CA013330).
Authorship
Contributions D.F., T.A., and I. Akhmetzyanova conceived, planned, conducted, and analyzed all the experiments; T.A. conducted all revision experiments; D.F. and I. Akhmetzyanova wrote the manuscript; P.G., A.T., I.D., I. Aifantis, D.Z., and X.Z. contributed to analysis and interpretation of RNA-seq experiments; M.T. contributed key biological materials and technical support; and all authors read and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: David Fooksman, Albert Einstein College of Medicine, 1300 Morris Park Ave, Bronx, NY 10461; e-mail: david.fooksman@einsteinmed.org.

