

Key Points
After 39.9 months of follow-up, time-to-event analyses were comparable between CT-P10 and rituximab in advanced-stage FL.
CT-P10 was well tolerated; the safety profile of CT-P10, including immunogenicity, was similar to that of rituximab over the study period.

Abstract
Rituximab biosimilars are a cornerstone of treatment of advanced-stage follicular lymphoma (FL). This double-blind, parallel-group, phase 3 trial randomized (1:1) adults (≥18 years) with stage III to IV indolent B-cell lymphoma, including grades 1 to 3a FL, to receive CT-P10 or rituximab (375 mg/m2 IV), with cyclophosphamide, vincristine, and prednisone, every 3 weeks for 8 cycles (induction period). Patients achieving complete response (CR), unconfirmed CR, or partial response (PR) received CT-P10 or rituximab maintenance for 2 years (375 mg/m2, every 8 weeks). Primary end points were previously reported, proving noninferiority of efficacy and pharmacokinetic equivalence of CT-P10 to rituximab. Secondary end points included overall response rate (PR+CR) during the induction period per 2007 International Working Group (IWG) criteria, survival analyses, and overall safety. Between 28 July 2014 and 29 December 2015, 140 patients were randomized (70 per group). Median follow-up was 39.9 months (interquartile range, 36.7-43.5). Per 1999 IWG criteria, 4-year Kaplan-Meier estimates (95% confidence interval [CI]) for CT-P10 and rituximab were 61% (47% to 73%) and 55% (36% to 70%) for progression-free survival (hazard ratio, 1.33 [95% CI, 0.67-2.63]; P=.409), respectively, and 88% (77% to 94%) and 93% (83% to 97%) for overall survival (5.29 [0.84-33.53]; P=.077). Overall, 90% (CT-P10) and 86% (rituximab) of patients experienced treatment-emergent adverse events. Long-term safety profiles were similar between groups. Findings confirm favorable outcomes for CT-P10–treated patients with advanced-stage FL and demonstrate comparable long-term efficacy and overall safety between CT-P10 and rituximab. This trial was registered at www.clinicaltrials.gov as #NCT02162771.
Introduction
Since its introduction, the anti-CD20 monoclonal antibody rituximab has changed the treatment landscape in follicular lymphoma (FL).1,2 Rituximab remains a cornerstone of therapy for this lymphoma subtype, particularly in combination with chemotherapy.1,2 Current treatment guidelines from the National Comprehensive Cancer Network (NCCN) suggest the combination of cyclophosphamide, vincristine, and prednisone (CVP) and rituximab as a preferred first-line treatment regimen, among others, for grade 1 to 2 advanced FL,3 while European Society for Medical Oncology guidelines recommend this regimen as one option for frontline treatment of high-tumor-burden stage III to IV disease.4 The NCCN guidelines note that rituximab biosimilars approved by the US Food and Drug Administration are an appropriate substitute for the reference product.3
CT-P10 (Truxima; Celltrion, Incheon, South Korea) was the first rituximab biosimilar to receive regulatory approval from authorities such as the European Medicines Agency and US Food and Drug Administration for indications including FL.5,6 Regulatory approval was supported by the analytical and nonclinical similarity of CT-P10 to both European Union– and United States–sourced rituximab.7 As with other biosimilars, CT-P10 uptake has been predicted to result in substantial cost savings, which could allow expanded patient access to rituximab treatment.8
This randomized, double-blind, phase 3 trial aimed to assess the noninferiority of efficacy and pharmacokinetic (PK) equivalence of CT-P10 to rituximab, in combination with CVP, in adults with newly diagnosed advanced-stage FL.9 The primary end points of the study have been previously reported; PK equivalence and noninferiority in efficacy, in terms of overall response rate (ORR), were demonstrated.9 This article reports the long-term secondary efficacy outcomes and updated safety findings for the overall study period.
Methods
Study design, patients, and procedures
This was a global, phase 3, randomized, double-blind, parallel-group, active-controlled study (www.clinicaltrials.gov #NCT02162771) with patients randomized at 65 centers (including one Good Clinical Practice noncompliant center, for which patients were excluded from all analysis populations). The study design and eligibility criteria have been previously published, together with the study protocol.9
Briefly, adult patients (age ≥18 years) with previously untreated, histologically confirmed, CD20-positive, grade 1 to 3a, advanced (Ann Arbor stage III-IV) FL were randomized (1:1) to receive CT-P10 or United States–sourced rituximab (Rituxan; Genentech, South San Francisco, CA). During the induction period, patients received 8 cycles of CT-P10 or rituximab (375 mg/m2 IV, diluted into 500 mL of normal saline, on day 1 of each 21-day cycle) with cyclophosphamide (750 mg/m2 IV on day 1), vincristine (1.4 mg/m2 IV [maximum 2 mg] on day 1), and prednisone (40 mg/m2 orally on days 1-5). Patients with complete response (CR), unconfirmed CR (CRu), or partial response (PR) after week 24 of the induction period continued to receive 375 mg/m2 IV CT-P10 or rituximab monotherapy once every 2 months for up to 2 years (maintenance period). Patients with no response or with disease progression did not receive further study treatment and discontinued the study. The initial infusion rate for CT-P10 or rituximab was 50 mg/h or 100 mg/h for cycle 1 of the induction and maintenance periods, respectively. This rate was increased by 50 mg/h every 30 minutes to a maximum of 400 mg/h in the absence of infusion toxicity. In subsequent cycles, infusions were initiated at 100 mg/h, and rates increased in the same way. Antipyretic, antihistamine, and glucocorticoid premedication was administered 30 minutes before each study drug infusion throughout the study. In addition, any medications that were appropriate and part of the study center practice, including antiemetics, hydration, or antacids, could be administered as premedication.
Follow-up continued at 3-month intervals for disease status until treatment with new anticancer therapy or disease progression and then at 6-month intervals until death or 3 years from the first day of cycle 1 for the induction period for the last patient.
The study was conducted in accordance with the Declaration of Helsinki, the protocol was approved by ethics committees at each center, and all patients provided written informed consent.9
Outcomes
Time-to-event analyses were secondary efficacy end points, comprising progression-free survival (PFS), time to progression (TTP), time to treatment failure (TTF), response duration, disease-free survival (DFS), and overall survival (OS). Time-to-event analyses were conducted using 1999 International Working Group (IWG) criteria10 and 2007 IWG criteria.11 PFS was defined as the interval between randomization and disease progression or death from any cause, whichever occurred first, whereas TTP was defined as the interval between randomization and disease progression or death as a result of lymphoma, whichever occurred first. TTF was defined as the interval between randomization and discontinuation of study treatment of any reason. Response duration was defined as the interval between the first time response criteria were met (CR, CRu, or PR) and first documentation of disease relapse or progression, whereas DFS was defined as the interval between first attained CR or CRu to disease recurrence or death as a result of lymphoma or acute toxicity of treatment, whichever occurred first. OS was defined as the interval between randomization and death from any cause. A post hoc analysis was conducted for progression of disease within 24 months (POD24), defined as time from first therapy to first documented progression, using 1999 IWG criteria. Another secondary efficacy end point was ORR (the proportion of patients who achieved CR or PR) during the induction period according to the 2007 IWG criteria. This assessment was conducted for patients who underwent positron emission tomography (PET) or PET with computed tomography (CT) per investigator’s discretion. Safety, including adverse events (AEs), and immunogenicity were also assessed over the study period.9 For immunogenicity testing, antidrug antibodies were measured using an enhanced chemiluminescence immunoassay method. A complement-dependent cytotoxicity assay (developed by Celltrion) was used to measure neutralizing antibodies (NAbs).
Statistical analyses
Details regarding the sample size have been reported.9 Secondary efficacy outcomes were evaluated in the intent-to-treat (ITT) population. For time-to-event analyses, the numbers of events and censored patients were summarized by treatment group, and survival rates (with corresponding 95% confidence intervals [CIs]) were estimated using the Kaplan-Meier method. Hazard ratios (HRs; with corresponding 95% CIs and P values) were estimated as the risk of event for CT-P10 over rituximab and were generated using a Cox proportional-hazards model with treatment as the main effect, adjusted for country, sex, race, Eastern Cooperative Oncology Group performance status, and baseline Follicular Lymphoma International Prognostic Index score (0-2 vs 3-5). Cumulative incidence function estimates for POD24 (with corresponding 95% CIs) were estimated in the ITT population; HRs with Gray’s test P values were estimated as for the time-to-event analyses. Safety analyses were conducted in the safety population, which comprised all randomized patients who had received at least 1 dose of study drug (CT-P10 or rituximab).
Role of the funding source
The sponsor was involved in conception and design of the study and in data collection, analysis, and interpretation. All authors, including employees of the sponsor, participated in article development, and all authors had full access to the complete clinical data set upon request. SJ.L., SH.K., and KY.A. had access to the raw data. The corresponding author had final responsibility for the decision to submit for publication.
Results
Patients were enrolled between 28 July 2014 and 29 December 2015,9 and the last follow-up visit was on 29 December 2018. Among 184 individuals screened for eligibility, 140 patients were randomly assigned to receive either CT-P10 (n = 70; 50%) or rituximab (n = 70; 50%) in combination with CVP (Figure 1). As previously reported,9 baseline patient demographics and disease characteristics were generally similar between groups, although a greater proportion of the CT-P10 group had bone marrow involvement and Ann Arbor stage IV FL vs the rituximab group.
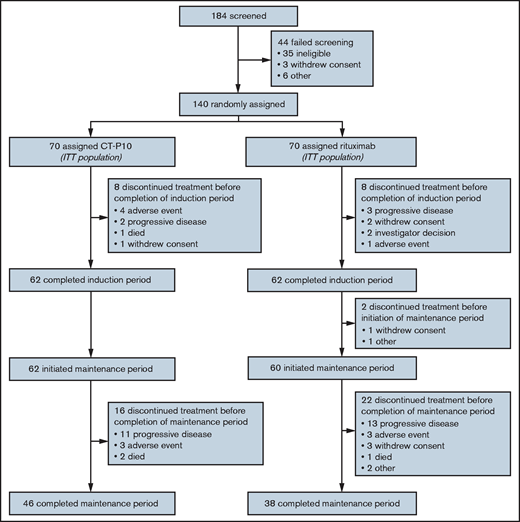
Of the 140 patients randomized, 62 (89%) from each of the CT-P10 and rituximab groups completed the induction period. All patients who completed the induction period started maintenance therapy, apart from 2 patients in the rituximab group. At study completion, 46 (66%) of 70 patients in the CT-P10 group and 38 (54%) of 70 patients in the rituximab group completed the full study treatment, receiving 12 cycles of maintenance therapy during the 2 years following the induction period. During the overall study period, the most frequent reason for treatment discontinuation was disease progression in both groups (CT-P10: 19% [13/70]; rituximab: 23% [16/70]).
Overall, the median follow-up duration was 39.9 months (interquartile range, 36.7-43.5). According to 1999 IWG criteria, median TTF was not reached in the CT-P10 group and was 41.9 months (95% CI, 22.1-not estimable) in the rituximab group (ITT population). Other than for TTF, the median was not reached in either treatment group in the ITT population for any time-to-event parameter owing to an insufficient number of events. However, Kaplan-Meier estimates were similar between treatment groups for all time-to-event parameters (Figure 2 and Table 1). For response duration, there were 67 and 61 evaluable patients in the CT-P10 and rituximab groups, respectively; most patients achieved a sustained response (CR, CRu, or PR), with proportions similar between treatment groups. In addition, post hoc analyses using Cox proportional-hazards models found no evidence of a difference in HR between treatment groups for each time-to-event parameter (1999 IWG criteria; ITT population; Figure 2). For time-to-event analyses according to 2007 IWG criteria, the number of evaluable patients was limited; however, there were no notable differences between groups (data not shown). In addition, the cumulative incidence of disease progression within 24 months was similar for both groups, providing further support for the comparability of CT-P10 and rituximab efficacy over the long term (Figure 3). A post hoc analysis showed that POD24 was observed in 21% (15/70) and 20% (14/70) of patients in CT-P10 and rituximab groups, respectively. Due to the very limited number of deaths, no clinically meaningful association between POD24 and OS was observed in this study (supplemental Figure 1).
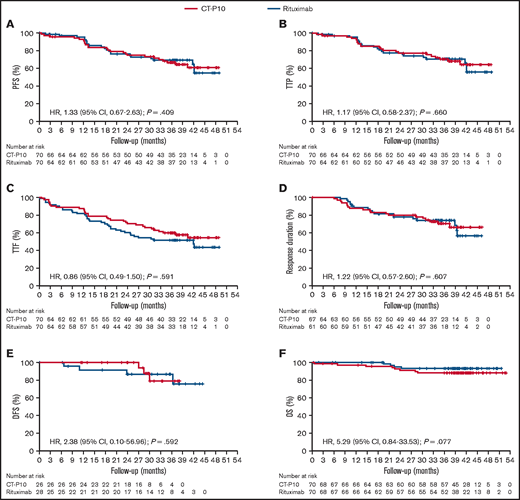
Kaplan-Meier curves for time-to-event analyses according to 1999 IWG criteria (ITT population). (A) PFS. (B) TTP. (C) TTF. (D) Response duration. (E) DFS. (F) OS.
Kaplan-Meier curves for time-to-event analyses according to 1999 IWG criteria (ITT population). (A) PFS. (B) TTP. (C) TTF. (D) Response duration. (E) DFS. (F) OS.
Kaplan-Meier estimates for time-to-event parameters according to 1999 IWG criteria (ITT population)
| Efficacy result, Kaplan-Meier estimate (95% CI) | 3-y follow-up | 4-y follow-up* | ||
|---|---|---|---|---|
| CT-P10 (n = 70) | Rituximab (n = 70) | CT-P10 (n = 70) | Rituximab (n = 70) | |
| PFS | 67% (54-77) | 69% (56-79) | 61% (47-73) | 55% (36-70) |
| TTP | 70% (57-80) | 71% (57-80) | 64% (49-76) | 56% (37-71) |
| TTF | 60% (47-70) | 51% (39-62) | 54% (41-66) | 44% (29-57) |
| Response duration | 70% (57-80) | 74% (60-83) | 66% (51-78) | 56% (35-73) |
| DFS | 79% (47-93) | 87% (65-96) | NE | NE |
| OS | 88% (77-94) | 93% (83-97) | 88% (77-94) | 93% (83-97) |
| Efficacy result, Kaplan-Meier estimate (95% CI) | 3-y follow-up | 4-y follow-up* | ||
|---|---|---|---|---|
| CT-P10 (n = 70) | Rituximab (n = 70) | CT-P10 (n = 70) | Rituximab (n = 70) | |
| PFS | 67% (54-77) | 69% (56-79) | 61% (47-73) | 55% (36-70) |
| TTP | 70% (57-80) | 71% (57-80) | 64% (49-76) | 56% (37-71) |
| TTF | 60% (47-70) | 51% (39-62) | 54% (41-66) | 44% (29-57) |
| Response duration | 70% (57-80) | 74% (60-83) | 66% (51-78) | 56% (35-73) |
| DFS | 79% (47-93) | 87% (65-96) | NE | NE |
| OS | 88% (77-94) | 93% (83-97) | 88% (77-94) | 93% (83-97) |
NE, not estimable.
3.5 y for response duration (last estimable time point in both treatment groups).
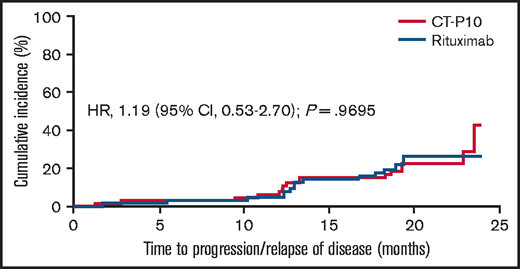
Cumulative incidence of disease progression per 1999 IWG criteria (ITT population).
Cumulative incidence of disease progression per 1999 IWG criteria (ITT population).
There were no notable differences between treatment groups in ORR by 2007 IWG criteria during the induction period, although the number of evaluable patients was limited (supplemental Table 1).
Exposure to CT-P10 or rituximab was similar between groups during the induction and maintenance periods: mean (standard deviation) relative doses were 97.7% (4.4) and 98.3% (2.7) for CT-P10 and rituximab groups, respectively, during the induction period; corresponding doses during the maintenance period were 101.0% (3.1) and 100.1% (3.7).
Overall, 63 (90%) and 60 (86%) patients experienced treatment-emergent AEs (TEAEs) in the CT-P10 and rituximab groups, respectively (Table 2). TEAEs considered by the investigator to be related to study drug were reported for 40 (57%) and 38 (54%) patients in the CT-P10 and rituximab groups, respectively. Correspondingly, during the maintenance period, 37 (60%) and 38 (63%) patients experienced TEAEs (supplemental Table 2). Over the study period, infusion-related reactions (IRRs) and neutropenia were the most frequent TEAEs in total (Table 3). Febrile neutropenia was reported by a similar proportion of patients in each treatment group during the overall study period (CT-P10, 3%; rituximab, 4%).
Summary of TEAEs during the overall study period (safety population)
| CT-P10 (n = 70) | Rituximab (n = 70) | |
|---|---|---|
| Patients with ≥1 TEAE | ||
| Any TEAE | 63 (90) | 60 (86) |
| Study drug related | 40 (57) | 38 (54) |
| Grade ≥4 TEAE | 12 (17) | 7 (10) |
| Study drug related | 5 (7) | 5 (7) |
| TESAE | 24 (34) | 13 (19) |
| Study drug related | 7 (10) | 6 (9) |
| Patients with ≥1 TEAE due to IRR | 16 (23) | 19 (27) |
| Patients with ≥1 TEAE due to infection | 35 (50) | 32 (46) |
| Patients with ≥1 TEAE leading to treatment discontinuation | 10 (14) | 5 (7) |
| Study drug related | 3 (4) | 3 (4) |
| CT-P10 (n = 70) | Rituximab (n = 70) | |
|---|---|---|
| Patients with ≥1 TEAE | ||
| Any TEAE | 63 (90) | 60 (86) |
| Study drug related | 40 (57) | 38 (54) |
| Grade ≥4 TEAE | 12 (17) | 7 (10) |
| Study drug related | 5 (7) | 5 (7) |
| TESAE | 24 (34) | 13 (19) |
| Study drug related | 7 (10) | 6 (9) |
| Patients with ≥1 TEAE due to IRR | 16 (23) | 19 (27) |
| Patients with ≥1 TEAE due to infection | 35 (50) | 32 (46) |
| Patients with ≥1 TEAE leading to treatment discontinuation | 10 (14) | 5 (7) |
| Study drug related | 3 (4) | 3 (4) |
Data are n (%) of patients.
Summary of TEAEs reported for >10% patients in either treatment group (safety population)
| Period: Preferred term | CT-P10 | Rituximab | ||||
|---|---|---|---|---|---|---|
| Grade 1 or 2 | Grade 3 | Grade 4 | Grade 1 or 2 | Grade 3 | Grade 4 | |
| Overall study period | n = 70 | n = 70 | ||||
| Abdominal pain | 7 (10) | 1 (1) | 0 | 11 (16) | 0 | 0 |
| Alopecia | 10 (14) | 0 | 0 | 5 (7) | 0 | 0 |
| Asthenia | 5 (7) | 0 | 0 | 8 (11) | 0 | 0 |
| Back pain | 2 (3) | 0 | 0 | 12 (17) | 0 | 0 |
| Constipation | 12 (17) | 0 | 0 | 10 (14) | 0 | 0 |
| Diarrhea | 6 (9) | 0 | 0 | 7 (10) | 1 (1) | 0 |
| Fatigue | 6 (9) | 0 | 0 | 7 (10) | 1 (1) | 0 |
| Infusion-related reaction | 14 (20) | 2 (3) | 0 | 19 (27) | 0 | 0 |
| Nausea | 9 (13) | 0 | 0 | 7 (10) | 0 | 0 |
| Neuropathy peripheral | 10 (14) | 0 | 0 | 11 (16) | 1 (1) | 0 |
| Neutropenia | 6 (9) | 14 (20) | 8 (11) | 8 (11) | 7 (10) | 5 (7) |
| Paresthesia | 2 (3) | 1 (1) | 0 | 8 (11) | 0 | 0 |
| Upper respiratory tract infection | 14 (20) | 0 | 0 | 15 (21) | 3 (4) | 0 |
| Maintenance study period | n = 62 | n = 60 | ||||
| Neutropenia | 2 (3) | 1 (2) | 3 (5) | 3 (5) | 3 (5) | 1 (2) |
| Upper respiratory tract infection | 11 (18) | 0 | 0 | 7 (12) | 3 (5) | 0 |
| Period: Preferred term | CT-P10 | Rituximab | ||||
|---|---|---|---|---|---|---|
| Grade 1 or 2 | Grade 3 | Grade 4 | Grade 1 or 2 | Grade 3 | Grade 4 | |
| Overall study period | n = 70 | n = 70 | ||||
| Abdominal pain | 7 (10) | 1 (1) | 0 | 11 (16) | 0 | 0 |
| Alopecia | 10 (14) | 0 | 0 | 5 (7) | 0 | 0 |
| Asthenia | 5 (7) | 0 | 0 | 8 (11) | 0 | 0 |
| Back pain | 2 (3) | 0 | 0 | 12 (17) | 0 | 0 |
| Constipation | 12 (17) | 0 | 0 | 10 (14) | 0 | 0 |
| Diarrhea | 6 (9) | 0 | 0 | 7 (10) | 1 (1) | 0 |
| Fatigue | 6 (9) | 0 | 0 | 7 (10) | 1 (1) | 0 |
| Infusion-related reaction | 14 (20) | 2 (3) | 0 | 19 (27) | 0 | 0 |
| Nausea | 9 (13) | 0 | 0 | 7 (10) | 0 | 0 |
| Neuropathy peripheral | 10 (14) | 0 | 0 | 11 (16) | 1 (1) | 0 |
| Neutropenia | 6 (9) | 14 (20) | 8 (11) | 8 (11) | 7 (10) | 5 (7) |
| Paresthesia | 2 (3) | 1 (1) | 0 | 8 (11) | 0 | 0 |
| Upper respiratory tract infection | 14 (20) | 0 | 0 | 15 (21) | 3 (4) | 0 |
| Maintenance study period | n = 62 | n = 60 | ||||
| Neutropenia | 2 (3) | 1 (2) | 3 (5) | 3 (5) | 3 (5) | 1 (2) |
| Upper respiratory tract infection | 11 (18) | 0 | 0 | 7 (12) | 3 (5) | 0 |
Data are n (%) of patients.
During the overall study period, 24 (34%) and 13 (19%) patients in the CT-P10 and rituximab groups, respectively, experienced treatment-emergent serious AEs (TESAEs) (Table 2). While more patients in the CT-P10 group than the rituximab group experienced TESAEs, the number of patients who experienced study drug–related TESAEs was similar between the groups (CT-P10: 7 [10%] patients; rituximab: 6 [9%] patients). After the previous report,9 11 TESAEs were reported in 10 (16%) patients treated with CT-P10 during the maintenance period, while 10 TESAEs were reported in 5 (8%) patients treated with rituximab (supplemental Table 2). During the maintenance period, study drug–related TESAEs were reported in 1 (2%) and 2 (3%) patients in the CT-P10 and rituximab groups, respectively. There were no clinically notable changes in clinical laboratory parameters from baseline in both treatment groups.
In terms of AEs of special interest, IRRs were reported in 16 (23%) and 19 (27%) patients in the CT-P10 and rituximab groups, respectively, during the study (Table 2). All TEAEs due to IRR were grade 1 or 2 in intensity, other than 3 grade 3 events that occurred during the induction period. Infections were experienced by 35 (50%) and 32 (46%) patients in the CT-P10 and rituximab groups, respectively. As previously reported, tuberculosis occurred in 1 (1%) patient in the rituximab group with a history of the disease, which resulted in permanent discontinuation of study treatment.9 During the maintenance period, 1 (1%) patient in each treatment group had abnormal, clinically significant findings on tuberculosis assessment. These findings were due to progressive disease with new pulmonary lesions, leading to treatment discontinuation (rituximab group) and bronchitis (CT-P10 group; patient completed study treatment after antibiotic therapy). No cases of progressive multifocal leukoencephalopathy or pregnancies were reported.
Overall, 5 (4%) patients died due to TEAEs. As previously reported, 1 (1%) patient in the CT-P10 group experienced unconfirmed tumor lysis syndrome.9 During the maintenance period, 2 (3%) patients in the CT-P10 group died due to TEAEs (hepatocellular carcinoma and respiratory failure) and 1 (1%) patient in the rituximab group died due to acute respiratory distress syndrome. All of these TEAEs were considered unrelated to study drug. One (1%) patient in the CT-P10 group died during the follow-up period due to a TEAE of gastric adenocarcinoma (considered unrelated to study drug), which occurred during the maintenance period.
Overall, 7 patients (CT-P10, 3 [4%] patients; rituximab, 4 [6%] patients) had at least 1 positive result for antidrug antibody tests at posttreatment visits during the overall study period. Among the 7 patients, 4 patients (2 [3%] patients in each treatment group) had positive NAb results, while the other 3 patients (CT-P10 1 [1%] patient; rituximab, 2 [3%] patients) had negative NAb results at posttreatment visits. The 4 patients who developed NAbs did so during the induction study period; there were no positive NAb results during the maintenance period.
Discussion
This report summarizes the efficacy and safety of CT-P10, as compared with rituximab, in combination with standard CVP chemotherapy in a large, international, prospective, randomized study of adults with newly diagnosed advanced-stage FL. After a median follow-up of 39.9 months overall, this report clearly demonstrates that the efficacy and safety findings for CT-P10 are robust over the long term. These data establish the noninferiority of CT-P10 and rituximab in the treatment of advanced-stage FL, demonstrating comparability in ORR during the induction period, as well as similar findings for time-to-event analyses and overall safety profiles. These findings build on the conclusions of PK equivalence and noninferior efficacy previously drawn for the primary end points of the study.9
To provide further support for noninferiority in efficacy between CT-P10 and rituximab, secondary efficacy analyses were planned that would be evaluated using the 2007 IWG criteria for patients who underwent PET or PET-CT imaging. However, the number of evaluable patients by 2007 IWG criteria was low, since few investigators chose to use PET or PET-CT imaging. Contributing factors may include the lack of established use of PET imaging (outside clinical trial contexts) and the partial reimbursement of PET imaging for patients with FL in some participating countries. Nevertheless, in this analysis, the ORR during the induction period according to 2007 IWG criteria (CT-P10, 93%; rituximab, 94%) was similar to the ORR according to 1999 IWG criteria reported in the primary publication (CT-P10, 97%; rituximab, 93%).9 The ORR by 2007 IWG criteria in the present study (CT-P10, 93%; rituximab, 94%) was also similar to the ORR of 97% (assessed by CT scans per IWG criteria) reported in a study evaluating rituximab and CVP (R-CVP) induction treatment followed by maintenance rituximab in patients with previously untreated indolent non-Hodgkin lymphoma.12
Assessment methods and treatment regimens vary between studies, but findings for time-to-event analyses in our study were generally comparable with previous reports in patients with advanced-stage FL who had been treated with induction R-CVP followed by maintenance rituximab. The estimated 3-year PFS rates were 77% in 2 previous studies,13,14 compared with 67% (CT-P10) and 69% (rituximab) in ours. In addition, the estimated 3-year OS of 97% reported previously14 was comparable to our findings of 88% (CT-P10) and 93% (rituximab). Our findings were also similar to the estimated 3-year PFS (64%) and OS (91%) reported for patients with low-grade FL who had received induction CVP followed by maintenance rituximab in another study.15 In addition, the 4-year OS of 91% in the HUSOM study, which evaluated maintenance rituximab treatment in patients with FL who had responded to induction therapy with R-CVP or rituximab with cyclophosphamide, doxorubicin, vincristine, and prednisone,16 was also similar to our findings. We also conducted a post hoc analysis of POD24, which is predictive of a worse OS.17 For this study, no clinically meaningful results were obtained in the analysis of the association of POD24 with OS, since few deaths occurred during the study. However, the incidence of POD24 was similar between groups.
The proportion of patients experiencing TEAEs during the maintenance period was lower than reported after the induction period.9 This might have been related to the CVP treatment administered during the induction period; rituximab has previously been reported to not significantly add to the toxicity of CVP.18 Overall, the proportion of patients experiencing TEAEs (CT-P10, 90%; rituximab, 86%) was comparable to that in the ASSIST-FL study, where 93% and 91% of patients receiving GP2013 and rituximab, respectively, reported AEs.19 Notably, the proportion of patients experiencing grade 3-4 TEAEs of neutropenia differed between treatment groups during the overall study period (CT-P10, 31%; rituximab, 17%), which was also reported during the induction period.9 As discussed in the primary publication, this imbalance may have been related to an uneven distribution of baseline risk factors of bone marrow involvement (CT-P10, 64%; rituximab, 47%) and advanced disease stage (Ann Arbor stage IV: CT-P10, 70%; rituximab, 49%) between treatment groups.9 Notably, the proportion of patients with febrile neutropenia was similar between treatment groups during the overall study period.
Because of an insufficient number of events, medians were not reached for most time-to-event parameters, and, as noted previously, the study was not powered to compare survival parameters between treatment groups.9 However, this study provides the longest-term follow-up data for CT-P10 treatment in advanced-stage FL available to date, and median follow-up duration was similar between treatment groups. The conclusion of comparability between CT-P10 and rituximab is in keeping with the findings of a recent phase 3 study comparing rituximab and CT-P10 monotherapy in patients with low-tumor-burden FL.20
In summary, the data from this study in terms of ORR, time-to-event findings, and long-term safety profile provide additional evidence to support the clinical comparability of CT-P10 and rituximab in patients with previously untreated advanced-stage FL.
Acknowledgments
The authors thank all study investigators, staff, and patients who contributed to this study.
This work was supported by Celltrion. Medical writing support (including development of a draft outline and subsequent drafts in consultation with the authors, assembling tables and figures, collating author comments, copyediting, fact checking, and referencing) was provided by Beatrice Tyrrell (Aspire Scientific, Bollington, United Kingdom) and funded by Celltrion.
Authorship
Contribution: C.B., L.K., W.-S.K., SJ.L., SH.K., KY.A., and M.O. conceived and designed the study, and analyzed and interpreted the data; W.J., J.-M.S., E.Z., J.S.K., J.-A.H.-R., A.P., M.V., and R.N. collected the data; C.B. and M.O. were responsible for writing the first version of the manuscript; all authors reviewed drafts of the manuscript and approved the final version; and all authors had access to the complete clinical data set.
Conflict-of-interest disclosure: C.B. reports research funding from Bayer, Celltrion, Janssen, MSD, and Roche and honoraria from AbbVie, Bayer, BeiGene, Celltrion, Hexal, Janssen, and Roche. W.J. reports grants from Celltrion, Roche, and Sandoz-Novartis. J.-M.S. reports honoraria for advisory and speaker roles from Celgene, Gilead, Incyte, Janssen, Novartis, Roche, and Takeda outside the submitted work. J.-A.H.-R. reports honoraria for advisory and speaker roles from AbbVie, AstraZeneca, BeiGene, BMS-Celgene, Gilead, GSK, Janssen, Jazz-Pharmaceuticals, Novartis, Roche, Rovi, Sanofi, and Takeda outside the submitted work. L.K. reports personal fees from Celltrion and Celltrion Healthcare during the conduct of this study. W.-S.K. reports research funding from Celltrion, Johnson & Johnson, Kyowa-Kirin, Pfizer, Roche, Sanofi, and Takeda. SJ.L. is an employee of Celltrion, receiving salary and stock options. SH.K. and KY.A. are employees of Celltrion. M.O. reports personal fees from Celltrion during the conduct of this study and personal fees from Celgene, Celltrion, Chugai, Daiichi Sankyo, Denovo Biopharma, Eisai, Meiji Seika Pharma, Mundipharma, SymBio, and Verastem outside the submitted work. The remaining authors declare no competing financial interests.
Correspondence: Michinori Ogura, Department of Hematology and Oncology, Kasugai Municipal Hospital, 1-1-1 Takakicho Kasugai, Aichi 486-8510, Japan; e-mail: magnoliamic@me.com.

