

Key Points
Norbin suppresses murine innate immunity to pneumococcal infection by limiting the role of neutrophils in bacterial clearance.
Norbin suppresses neutrophil bactericidal responses, GPCR trafficking, and signaling via Rac and Erk.

Abstract
Streptococcal pneumonia is a worldwide health problem that kills ∼2 million people each year, particularly young children, the elderly, and immunosuppressed individuals. Alveolar macrophages and neutrophils provide the early innate immune response to clear pneumococcus from infected lungs. However, the level of neutrophil involvement is context dependent, both in humans and in mouse models of the disease, influenced by factors such as bacterial load, age, and coinfections. Here, we show that the G protein–coupled receptor (GPCR) adaptor protein norbin (neurochondrin, NCDN), which was hitherto known as a regulator of neuronal function, is a suppressor of neutrophil-mediated innate immunity. Myeloid norbin deficiency improved the immunity of mice to pneumococcal infection by increasing the involvement of neutrophils in clearing the bacteria, without affecting neutrophil recruitment or causing autoinflammation. It also improved immunity during Escherichia coli–induced septic peritonitis. It increased the responsiveness of neutrophils to a range of stimuli, promoting their ability to kill bacteria in a reactive oxygen species–dependent manner, enhancing degranulation, phagocytosis, and the production of reactive oxygen species and neutrophil extracellular traps, raising the cell surface levels of selected GPCRs, and increasing GPCR-dependent Rac and Erk signaling. The Rac guanine-nucleotide exchange factor Prex1, a known effector of norbin, was dispensable for most of these effects, which suggested that norbin controls additional downstream targets. We identified the Rac guanine-nucleotide exchange factor Vav as one of these effectors. In summary, our study presents the GPCR adaptor protein norbin as an immune suppressor that limits the ability of neutrophils to clear bacterial infections.
Introduction
Streptococcus pneumoniae (pneumococcus) infection is the leading cause of community-acquired pneumonia. It is particularly harmful to young children, the elderly, and immunocompromised individuals, resulting in high morbidity and mortality in both developing and industrialized countries. Approximately 2 million people die of pneumococcal infection worldwide each year.1 The early innate immune response to pneumococcal infection is provided by alveolar macrophages. Neutrophils become important in this early response when alveolar macrophages are overwhelmed by the severity of infection, comorbidities, coinfections, or immunosuppression.2
Neutrophil- and macrophage-mediated antibacterial immunity requires signaling through Rac small guanosine triphosphatases (GTPases) and Rac guanine-nucleotide exchange factors (Rac-GEFs), the guanine-nucleotide exchange factors that activate Rac. These proteins control the adhesion and migration of neutrophils and macrophages, as well as effector responses essential for immunity, including phagocytosis, degranulation, and the production of reactive oxygen species (ROS) and neutrophil extracellular traps (NETs).3 P-Rex1 is one of the Rac-GEFs controlling these responses, with Prex1-deficient mice exhibiting impaired neutrophil and macrophage migration, ROS production, and tissue recruitment.4-12
We identified a new regulator of P-Rex1, the adaptor protein norbin,13 an essential yet understudied cytosolic protein that is evolutionarily conserved but lacks homology to other proteins.14 Norbin is believed to be exclusively neuronal, except for low levels in chondrocytes and osteoblasts, and is recognized for its roles in the nervous system. Manipulation of norbin levels alters neuronal differentiation.15–17 General norbin deficiency in mice is embryonically lethal,18 but targeted deletion from selected types of neurons impairs adult neurogenesis, neuronal plasticity, and spatial learning, leading to epilepsy, schizophrenia, and depressive behaviors.19-21 These phenotypes may be pertinent to humans, as norbin levels are dysregulated in epilepsy and schizophrenia.22,23 Norbin is also a target antigen in autoimmune neurodegenerative diseases.24-27
Norbin binds directly to the intracellular tail of numerous G protein–coupled receptors (GPCRs).20,28,29 It can alter GPCR signaling and the steady-state cell surface level of GPCRs. However, the underlying mechanisms remain unclear. We have shown that norbin can bind P-Rex1, stimulate its catalytic Rac-GEF activity and membrane translocation, and induce P-Rex1/Rac–dependent cytoskeletal dynamics.13 We proposed that norbin promotes P-Rex1 function by bringing the Rac-GEF into proximity of Rac and its membrane-bound activators, PIP3 and Gβγ, thus facilitating Rac-dependent cell responses.
We found that norbin is not only expressed in neurons but also in myeloid cells (neutrophils and macrophages). Because norbin regulates P-Rex1, and P-Rex1 regulates neutrophil and macrophage responses, we decided to evaluate norbin function in myeloid cells. We generated mouse strains with myeloid norbin deficiency and combined norbin/Prex deficiency, expecting this to exacerbate the defects in neutrophil and macrophage responses seen in Prex1−/− mice. Surprisingly, we found that norbin deficiency improves neutrophil-mediated immunity, largely independently of P-Rex1.
Methods
Detailed protocols are provided in the supplemental Methods and supplemental Tables 1 and 2.
Mice
The NcdnΔmye mouse strain was generated by crossing Ncdnfl/fl19 with LysMCre,30 and the NcdnΔmyePrex1/2−/− strain was generated by crossing NcdnΔmye with Prex1/2‒/‒ (Prex1−/− Prex2−/−).31 Mice were sex- and age-matched for experiments. Animal breeding and experiments were performed with approval from the local ethical review body and the British Home Office.
S pneumoniae infection
Mice were infected intranasally with 2 × 106 colony-forming units (CFU) of S pneumoniae and bronchoalveolar lavage (BAL) and perfused lung homogenates analyzed essentially as described elsewhere.32 Where indicated, macrophages were depleted with clodronate liposomes33 and neutrophils with 1A8 antibody34 before pneumococcal infection. Histology and cytokine analysis were conducted as described elsewhere.9
Neutrophil responses
To measure bacterial killing, pathogens were incubated with purified neutrophils and CFU enumerated. ROS production was measured by using luminol chemiluminescence assay8; gelatinase degranulation by in-gel zymography35; adhesion, morphology, and phagocytosis by widefield imaging; and NETs by Sytox Green staining.36 Neutrophil surface levels of GPCRs and adhesion molecules were analyzed by flow cytometry9; integrin avidity by imaging37; Rac activity by Pak-CRIB pull down38; and Vav, Erk1/2, p38MAPK, Akt, and Jnk activities by western blotting.
Results
Myeloid norbin deficiency
To study myeloid norbin function, we generated NcdnΔmye mouse strain, which is norbin deficient in the myeloid lineage, by crossing Ncdnfl/fl,19 to LysMCre.30NcdnΔmye mice were also crossed to Prex1/2−/− (Prex1−/−Prex2−/−31) mice, which are deficient in Prex1 and its homolog Prex2, to generate NcdnΔmyePrex1/2−/− strain with combined norbin/Prex deficiency (Figure 1A). Western blotting of total lysates from resident peritoneal leukocytes (∼90% macrophages), peritonitis leukocytes (∼15% neutrophils, 75% macrophages), and mature bone marrow–derived neutrophils (98%) showed effective deletion of norbin (Figure 1B-C). Normal numbers of mature NcdnΔmye and NcdnΔmyePrex1/2−/− neutrophils with the characteristic segmented nuclear morphology, granule-derived cell density, and CD11bhi, Ly6Ghi surface marker expression were recovered from the bone marrow; normal numbers of neutrophils were present in the circulation (Figure 1D-G). Neutrophil development is thus normal in mice with myeloid norbin deficiency and combined norbin/Prex deficiency.
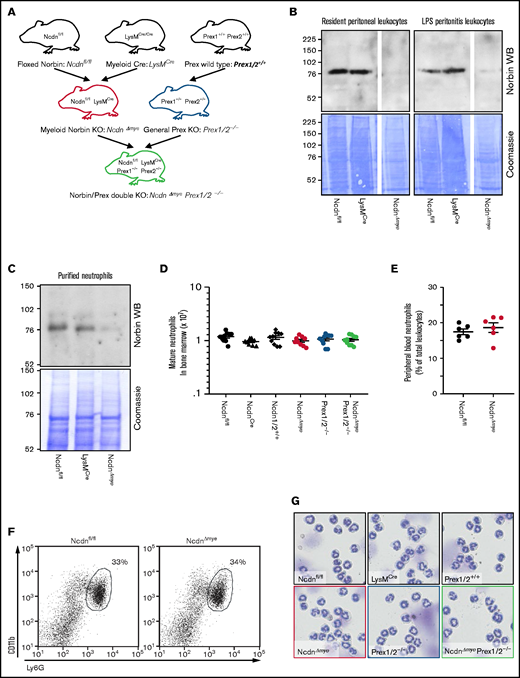
Neutrophildevelopment is normal in mice with myeloid norbin deficiency. (A) Schematic showing the generation of mouse strains with myeloid norbin deficiency (NcdnΔmye) and with combined norbin/Prex deficiency (NcdnΔmyePrex1/2−/−). Norbin deletion from resident peritoneal leukocytes and LPS-induced peritonitis leukocytes (B) and bone marrow–derived isolated neutrophils (C) of NcdnΔmye mice. Norbin western blots (WBs) and Coomassie staining of total cell lysates. (D) Quantification of mature bone marrow–derived neutrophils of the indicated genotypes. Data are mean ± standard error of the mean of 10 independent experiments. (E) Quantification of Ly6Ghi, CD11bhi neutrophils in peripheral blood as percentage of total leukocytes. Data are mean ± standard error of the mean; dots represent individual mice. (F) Representative flow cytometry plots showing expected numbers and marker protein levels of mature CD11bhi, Ly6Ghi neutrophils in bone marrow as percentage of total leukocytes. (G) Representative cytospin images (Diff-Quik stain) showing the characteristic nuclear morphology of mature isolated neutrophils as in panel D.
Neutrophildevelopment is normal in mice with myeloid norbin deficiency. (A) Schematic showing the generation of mouse strains with myeloid norbin deficiency (NcdnΔmye) and with combined norbin/Prex deficiency (NcdnΔmyePrex1/2−/−). Norbin deletion from resident peritoneal leukocytes and LPS-induced peritonitis leukocytes (B) and bone marrow–derived isolated neutrophils (C) of NcdnΔmye mice. Norbin western blots (WBs) and Coomassie staining of total cell lysates. (D) Quantification of mature bone marrow–derived neutrophils of the indicated genotypes. Data are mean ± standard error of the mean of 10 independent experiments. (E) Quantification of Ly6Ghi, CD11bhi neutrophils in peripheral blood as percentage of total leukocytes. Data are mean ± standard error of the mean; dots represent individual mice. (F) Representative flow cytometry plots showing expected numbers and marker protein levels of mature CD11bhi, Ly6Ghi neutrophils in bone marrow as percentage of total leukocytes. (G) Representative cytospin images (Diff-Quik stain) showing the characteristic nuclear morphology of mature isolated neutrophils as in panel D.
Myeloid norbin deficiency improves immunity against pneumococcal infection
To test the impact of myeloid norbin on immunity to pneumococcus, we subjected NcdnΔmye and NcdnΔmyePrex1/2−/− mice to intranasal infection with 2 × 106 CFU S pneumoniae.32 Bacterial clearance and leukocyte recruitment were measured at 6 hours and 18 hours after infection in BAL fluid and perfused lung homogenate to evaluate the airways and lung parenchyma, respectively. Pilot experiments with Ncdnfl/fl control mice showed that neutrophil recruitment increased between 6 and 18 hours, both in airways and parenchyma, whereas bacterial burden decreased sharply in the former but persisted longer in the latter (supplemental Figure 1). No obvious clinical signs were observed under these conditions. We compared NcdnΔmye mice vs Ncdnfl/fl and LysMCre control strains, and NcdnΔmyePrex1/2−/− mice vs Prex1/2+/+ and Prex1/2−/−.
Surprisingly, NcdnΔmye mice cleared an order of magnitude more bacteria from their airways within 6 hours than Ncdnfl/fl and LysMCre mice, despite normal neutrophil recruitment and alveolar macrophage numbers (Figure 2A). A similar increase in immunity was observed in the lung parenchyma, both at 6 and 18 hours (supplemental Figure 2). All strains had largely cleared the bacteria from their airways after 18 hours, except Prex1/2−/−, which retained significant bacterial burden. NcdnΔmyePrex1/2−/− mice were protected from this immunodeficiency, again without increased neutrophil or macrophage numbers (Figure 2B). Thus, norbin deletion increases immunity to pneumococcal infection and protects mice from immunodeficiency caused by lack of Prex.
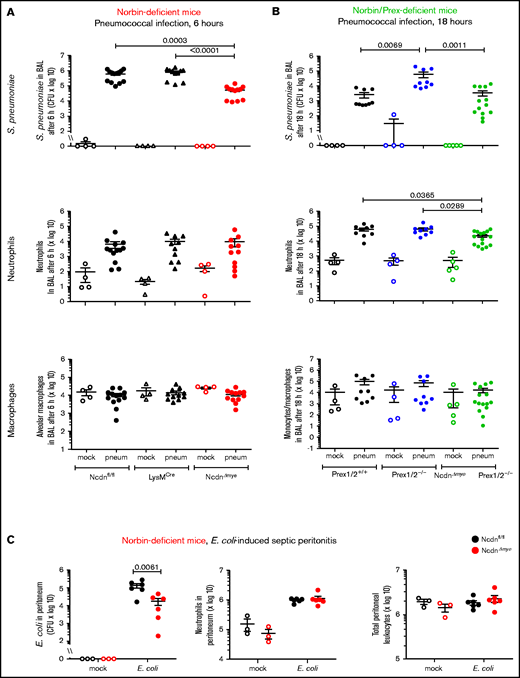
Myeloid norbin deficiency increases immunity during pneumococcal infection and septic peritonitis. Pneumococcal infection. Mice of the indicated genotypes were infected intranasally with 2 × 106S pneumoniae (filled symbols), or were mock-infected (open symbols), and culled after 6 hours (A) or after 18 hours (B). BALs were assessed for bacterial burden by quantification of CFU and for numbers of neutrophils (CD11bhi, Ly6Ghi), alveolar macrophages (CD11c+, SiglecF+), and monocytes/macrophages (CD11c+, SiglecF+; CD11b+, Ly6Glo; and CD11b+, Ly6C+) by flow cytometry. Data are mean ± standard error of the mean, pooled from 4 independent experiments for panel A or 5 for panel B; dots represent individual mice, typically 1 mock-infected and 2 to 3 infected mice per genotype per experiment. Statistics comprised Kruskal-Wallis analysis with Dunn’s multiple comparisons test. (C) Septic peritonitis. Mice were infected intraperitoneally with 1 × 105E coli (filled symbols), or were mock-infected (open symbols), and culled after 3 hours. Peritoneal lavages were assessed for bacterial burden by quantification of CFU and for numbers of neutrophils and total peritoneal leukocytes by using microscopy. Data are mean ± standard error of the mean, pooled from 3 independent experiments, with 1 mock-infected and 1 to 3 infected mice/genotype/experiment; dots represent individual mice. Statistics comprised two-way analysis of variance with Šidák’s multiple comparisons test.
Myeloid norbin deficiency increases immunity during pneumococcal infection and septic peritonitis. Pneumococcal infection. Mice of the indicated genotypes were infected intranasally with 2 × 106S pneumoniae (filled symbols), or were mock-infected (open symbols), and culled after 6 hours (A) or after 18 hours (B). BALs were assessed for bacterial burden by quantification of CFU and for numbers of neutrophils (CD11bhi, Ly6Ghi), alveolar macrophages (CD11c+, SiglecF+), and monocytes/macrophages (CD11c+, SiglecF+; CD11b+, Ly6Glo; and CD11b+, Ly6C+) by flow cytometry. Data are mean ± standard error of the mean, pooled from 4 independent experiments for panel A or 5 for panel B; dots represent individual mice, typically 1 mock-infected and 2 to 3 infected mice per genotype per experiment. Statistics comprised Kruskal-Wallis analysis with Dunn’s multiple comparisons test. (C) Septic peritonitis. Mice were infected intraperitoneally with 1 × 105E coli (filled symbols), or were mock-infected (open symbols), and culled after 3 hours. Peritoneal lavages were assessed for bacterial burden by quantification of CFU and for numbers of neutrophils and total peritoneal leukocytes by using microscopy. Data are mean ± standard error of the mean, pooled from 3 independent experiments, with 1 mock-infected and 1 to 3 infected mice/genotype/experiment; dots represent individual mice. Statistics comprised two-way analysis of variance with Šidák’s multiple comparisons test.
To test another type of infection, we induced peritonitis with pathogenic Escherichia coli O18:K1.39NcdnΔmye mice cleared an order of magnitude more E coli than Ncdnfl/fl mice, despite normal neutrophil recruitment and peritoneal leukocyte numbers (Figure 2C). Hence, myeloid norbin suppresses immunity against different bacteria and in different organs.
We also evaluated neutrophil recruitment in thioglycollate-induced aseptic peritonitis, which is Prex1 dependent.10 Peritoneal neutrophil recruitment was slightly elevated in NcdnΔmye compared with Ncdnfl/fl mice but not LysMCre mice, and thus considered normal overall (supplemental Figure 3A-C). Norbin deficiency did not overcome the requirement for Prex1, as recruitment was reduced in both Prex1/2−/− mice and NcdnΔmyePrex1/2−/− mice (supplemental Figure 3D). Hence, norbin is dispensable for neutrophil recruitment during both septic and aseptic inflammation.
Norbin thus plays an unexpected role as a suppressor of the early innate immune response to bacterial infection.
Improved immunity of norbin-deficient mice derives from neutrophils
We explored the cellular origin of the increased immunity, because both macrophages and neutrophils are involved in clearing pneumococcus, and the importance of neutrophils depends on factors such as bacterial load, age, and coinfections.2,40,41
To test macrophage involvement, we depleted monocytes/macrophages using clodronate liposomes33 before pneumococcal infection. This action reduced alveolar macrophage numbers in Ncdnfl/fl and NcdnΔmye mice by 81% and 75%, respectively, whereas neutrophil numbers were unaffected (Figure 3A). Macrophage depletion caused a 20-fold decrease in bacterial clearance, confirming that alveolar macrophages are essential for the early immune response to pneumococcus, as expected. However, the immunity of NcdnΔmye mice remained an order of magnitude better than that of Ncdnfl/fl mice. Hence, norbin is dispensable for macrophage-dependent immunity to pneumococcal infection.
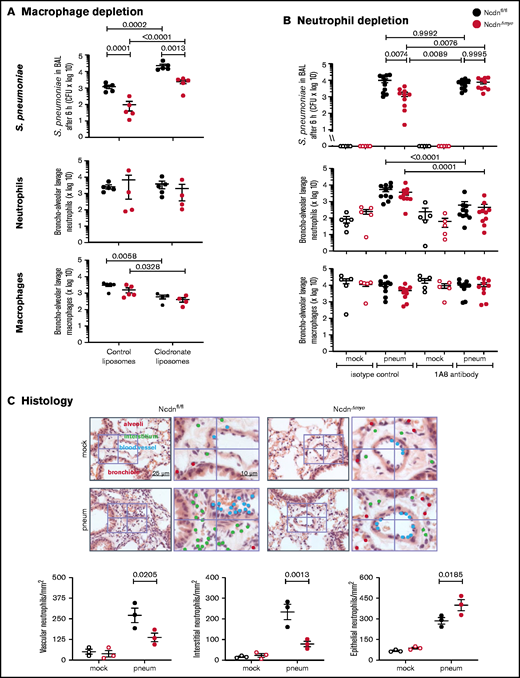
The increased immunity of norbin-deficient mice derives from neutrophils. (A) Macrophage depletion. Ncdnfl/fl and NcdnΔmye mice were treated intravenously with 100 μL and intranasally with 50 μL clodronate liposomes, or were mock-treated with control liposomes, at 72 hours before intranasal infection with 2 × 106S pneumoniae; they were culled 6 hours later and BAL performed. (B) Neutrophil depletion. Ncdnfl/fl and NcdnΔmye mice were treated with 1A8 monoclonal Ly6G antibody or isotype control immunoglobulin G at 24 hours and 0 hours before intranasal infection with 2 × 106S pneumoniae (filled symbols), or mock-infected (open symbols), culled 6 hours later, and BAL performed. BAL was analyzed for bacterial burden by quantification of CFU and for numbers of neutrophils (CD11bhi, CD24hi in panel A; CD11bhi, Ly6Ghi in panel B) and macrophages (CD11c+, SiglecF+ and CD11b+, CD24lo) by flow cytometry. Data in panel A are mean ± standard error of the mean pooled from 2 independent experiments; 2 to 3 mock-infected and 2 to 3 infected mice/genotype/experiment. Data in panel B are mean ± standard error of the mean pooled from 5 independent experiments; 1 mock-infected and 2 infected mice/genotype/experiment. Dots represent individual mice. Statistics comprised two-way analysis of variance with Šidák’s (panel A) or Tukey’s (panel B) multiple comparisons tests. (C) Histology. Ncdnfl/fl and NcdnΔmye mice were infected intranasally with 2 × 106S pneumoniae (filled symbols), or mock-infection (open symbols), culled 6 hours later, lungs fixed in formalin, and hematoxylin and eosin–stained sections analyzed for the presence of neutrophils according to their characteristic nuclear morphology. For each mouse, 60 fields of view of 40 000 μm2 from 6 areas distributed throughout the lung were analyzed. Left-hand panels show one representative field of view; right-hand panels are zoom-ins to show labeling of neutrophils within 10 μm of blood vessels or airway epithelium, or within the interstitium using different color dots. The quantification below shows mean ± standard error of the mean of 3 mice per group. Statistics comprised two-way analysis of variance with Šidák’s multiple comparisons test.
The increased immunity of norbin-deficient mice derives from neutrophils. (A) Macrophage depletion. Ncdnfl/fl and NcdnΔmye mice were treated intravenously with 100 μL and intranasally with 50 μL clodronate liposomes, or were mock-treated with control liposomes, at 72 hours before intranasal infection with 2 × 106S pneumoniae; they were culled 6 hours later and BAL performed. (B) Neutrophil depletion. Ncdnfl/fl and NcdnΔmye mice were treated with 1A8 monoclonal Ly6G antibody or isotype control immunoglobulin G at 24 hours and 0 hours before intranasal infection with 2 × 106S pneumoniae (filled symbols), or mock-infected (open symbols), culled 6 hours later, and BAL performed. BAL was analyzed for bacterial burden by quantification of CFU and for numbers of neutrophils (CD11bhi, CD24hi in panel A; CD11bhi, Ly6Ghi in panel B) and macrophages (CD11c+, SiglecF+ and CD11b+, CD24lo) by flow cytometry. Data in panel A are mean ± standard error of the mean pooled from 2 independent experiments; 2 to 3 mock-infected and 2 to 3 infected mice/genotype/experiment. Data in panel B are mean ± standard error of the mean pooled from 5 independent experiments; 1 mock-infected and 2 infected mice/genotype/experiment. Dots represent individual mice. Statistics comprised two-way analysis of variance with Šidák’s (panel A) or Tukey’s (panel B) multiple comparisons tests. (C) Histology. Ncdnfl/fl and NcdnΔmye mice were infected intranasally with 2 × 106S pneumoniae (filled symbols), or mock-infection (open symbols), culled 6 hours later, lungs fixed in formalin, and hematoxylin and eosin–stained sections analyzed for the presence of neutrophils according to their characteristic nuclear morphology. For each mouse, 60 fields of view of 40 000 μm2 from 6 areas distributed throughout the lung were analyzed. Left-hand panels show one representative field of view; right-hand panels are zoom-ins to show labeling of neutrophils within 10 μm of blood vessels or airway epithelium, or within the interstitium using different color dots. The quantification below shows mean ± standard error of the mean of 3 mice per group. Statistics comprised two-way analysis of variance with Šidák’s multiple comparisons test.
To test the role of neutrophils, we depleted neutrophils with 1A8 antibody34 before pneumococcal infection, which reduced neutrophil numbers in the peripheral blood of Ncdnfl/fl and Ncdn‒/‒ mice by 70% and 83%, respectively (supplemental Figure 4), and neutrophil recruitment into the airways by 89% and 88%; monocyte/macrophage numbers remained unaltered (Figure 3B). Ncdnfl/fl animals had a similar bacterial burden regardless of whether they were neutrophil depleted, which supports previous reports that in otherwise healthy adult mice, neutrophils are nonessential for clearing pneumococcus.2,41 However, the increased immunity of NcdnΔmye mice compared with Ncdnfl/fl mice was lost upon neutrophil depletion, showing that it derives from neutrophils.
To further characterize the immune response of NcdnΔmye mice, we tested chemokine/cytokine production and performed histologic analysis.9S pneumoniae–induced levels of tumor necrosis factor-α (TNF-α), CXCL1, and interleukin-6 in the lung were normal (supplemental Figure 5). Histologic analysis confirmed low inflammatory cells in the lungs of mock-infected mice and normal levels of pneumococcus-induced neutrophil recruitment overall, although the distribution of recruited neutrophils within lung tissue was altered. NcdnΔmye mice had fewer neutrophils in the vasculature and interstitium and a concomitant increase in airway infiltration (Figure 3C).
Together, these results showed that myeloid norbin deficiency enhances the immunity of mice to pneumococcus in a neutrophil-dependent manner by rendering neutrophils more important in the fight against this infection than they ordinarily are, without causing enhanced inflammatory states. Considering that neutrophil recruitment was unaffected, this scenario implied that antibacterial neutrophil responses may be increased.
Norbin-deficient neutrophils have an increased capacity to kill bacteria that is ROS dependent
We examined the ability of isolated neutrophils to kill bacteria. NcdnΔmye neutrophils killed more S pneumoniae or Staphylococcus aureus than Ncdnfl/fl and LysMCre control cells (Figure 4A-B). In contrast, Prex1/2−/− neutrophils killed fewer S pneumoniae than Prex1/2+/+ cells (Figure 4C). This was overcome by norbin deficiency, as NcdnΔmyePrex1/2−/− neutrophils showed the same killing capacity as NcdnΔmye cells. Hence, norbin deficiency enhances neutrophil-mediated bacterial killing, independently of Prex1.

Norbin-deficient neutrophils have an increased ability to kill bacteria, in a ROS-dependent manner, and increased ROS production in response to pathogens. Isolated neutrophils of the indicated genotypes were primed with 50 ng/mL TNF-α, 20 ng/mL GM-CSF before incubation with serum-opsonized S pneumoniae at a ratio of 1:1 (A) or 1:2 (C) for 15 minutes. Samples were sonicated in 0.05% saponin and serial dilutions cultured on blood agar to quantify CFU. Isolated neutrophils primed as in panel A were incubated with serum-opsonized S aureus at a ratio of 1:1 (B) or 1:2 (D) bacteria to neutrophils for 15 minutes. Panel D includes the indicated concentrations of the ROS inhibitor diphenyleneiodonium (DPI). CFU were determined as in panel A except that samples were cultured on luria broth agar. Data are mean ± standard error of the mean of 3 independent experiments for panel A, 5 to 9 for panel B, 4 for panel C, and 7 for panel D; each dot is the mean of 1 experiment performed in duplicate. The right-hand part of panel D shows samples without neutrophils. Statistics comprised one-way analysis of variance with Tukey’s multiple comparison test (panels A and B), paired two-tailed Student t test (panel C), and two-way analysis of variance with Šidák’s multiple comparisons test (panel D). (E) ROS production by isolated neutrophils from mice of the indicated genotypes, measured by using real-time chemiluminescence assay with luminol and horseradish peroxidase for extracellular and intracellular ROS, stimulated with serum-opsonized heat-inactivated S pneumoniae (200 bacteria/neutrophil), serum-opsonized live S aureus (5 bacteria/neutrophil), or zymosan (3 particles/neutrophil), as indicated, and quantified as area under the curve (AUC). Mock-stimulated ROS production was subtracted and is not shown for clarity. Data are mean ± standard error of the mean of the indicated numbers of independent experiments; each dot is the mean AUC from 1 experiment. Statistics comprised one-way analysis of variance with Holm-Šidák’s (S pneumoniae, S aureus) or Tukey’s (zymosan) multiple comparisons tests.
Norbin-deficient neutrophils have an increased ability to kill bacteria, in a ROS-dependent manner, and increased ROS production in response to pathogens. Isolated neutrophils of the indicated genotypes were primed with 50 ng/mL TNF-α, 20 ng/mL GM-CSF before incubation with serum-opsonized S pneumoniae at a ratio of 1:1 (A) or 1:2 (C) for 15 minutes. Samples were sonicated in 0.05% saponin and serial dilutions cultured on blood agar to quantify CFU. Isolated neutrophils primed as in panel A were incubated with serum-opsonized S aureus at a ratio of 1:1 (B) or 1:2 (D) bacteria to neutrophils for 15 minutes. Panel D includes the indicated concentrations of the ROS inhibitor diphenyleneiodonium (DPI). CFU were determined as in panel A except that samples were cultured on luria broth agar. Data are mean ± standard error of the mean of 3 independent experiments for panel A, 5 to 9 for panel B, 4 for panel C, and 7 for panel D; each dot is the mean of 1 experiment performed in duplicate. The right-hand part of panel D shows samples without neutrophils. Statistics comprised one-way analysis of variance with Tukey’s multiple comparison test (panels A and B), paired two-tailed Student t test (panel C), and two-way analysis of variance with Šidák’s multiple comparisons test (panel D). (E) ROS production by isolated neutrophils from mice of the indicated genotypes, measured by using real-time chemiluminescence assay with luminol and horseradish peroxidase for extracellular and intracellular ROS, stimulated with serum-opsonized heat-inactivated S pneumoniae (200 bacteria/neutrophil), serum-opsonized live S aureus (5 bacteria/neutrophil), or zymosan (3 particles/neutrophil), as indicated, and quantified as area under the curve (AUC). Mock-stimulated ROS production was subtracted and is not shown for clarity. Data are mean ± standard error of the mean of the indicated numbers of independent experiments; each dot is the mean AUC from 1 experiment. Statistics comprised one-way analysis of variance with Holm-Šidák’s (S pneumoniae, S aureus) or Tukey’s (zymosan) multiple comparisons tests.
To test whether the increased killing of S aureus by NcdnΔmye neutrophils is ROS dependent, the ROS inhibitor diphenyleneiodonium was included in killing assays. With increasing doses of diphenyleneiodonium, NcdnΔmye neutrophils lost their advantage over Ncdnfl/fl cells in killing S aureus (Figure 4D). Therefore, the increased capacity of norbin-deficient neutrophils for killing bacteria is ROS dependent, suggesting their ROS production may be enhanced.
Norbin deficiency increases ROS production
We evaluated pathogen-stimulated ROS production in NcdnΔmye and NcdnΔmyePrex1/2−/− neutrophils. Ncdn‒/‒ neutrophils produced significantly more ROS than Ncdnfl/fl in response to heat-inactivated S pneumoniae, live S aureus, or yeast-derived zymosan (Figure 4E). NcdnΔmyePrex1/2−/− neutrophils also produced more ROS than Prex1/2−/−cells in response to S pneumoniae and possibly S aureus but not zymosan. Hence, norbin-deficient cells have an increased ROS response to bacterial and fungal pathogens, with varying requirements for Prex1.
Because norbin regulates GPCR signaling,18,28,29 we assessed GPCR-mediated ROS production. Neutrophils were primed with TNF-α/granulocyte-macrophage colony-stimulating factor (GM-CSF) or lipopolysaccharide (LPS) to upregulate receptors and NADPH oxidase to the plasma membrane, and stimulated them with fMLP or C5a to activate FPR1/2 or C5aR1. Without priming, fMLP- and C5a-stimulated ROS production was negligible, and priming alone did not elicit ROS (not shown), indicating that norbin-deficient neutrophils are not preactivated. TNF-α/GM-CSF–primed, fMLP-stimulated ROS were increased in NcdnΔmye neutrophils and reduced both in Prex1/2−/− and NcdnΔmyePrex1/2−/− cells (supplemental Figure 6A). Similar effects were seen upon stimulation with C5a (supplemental Figure 6B). LPS-primed, fMLP-stimulated ROS were also increased in NcdnΔmye neutrophils, and reduced in Prex1/2−/− cells, but not in NcdnΔmyePrex1/2−/− cells (supplemental Figure 6C). Hence, norbin deficiency increases GPCR-dependent ROS production and overcomes the requirement for Prex1 during LPS priming but not TNF-α/GM-CSF priming, and thus the interdependence of norbin and Prex1 varies between signaling pathways.
Importantly, receptor-independent ROS production stimulated by phorbol 12‐myristate 13‐acetate was normal in all genotypes (supplemental Figure 6D). This finding means there is no change in overall NADPH oxidase capacity, and myeloid norbin deficiency affects upstream pathways rather than the NADPH oxidase itself.
In summary, norbin deficiency increases ROS production in response to a wide range of pathogens and signaling pathways. This increased ROS can explain, in part, the improved antibacterial capacity of norbin-deficient neutrophils. Overall, norbin has an opposite and more fundamental role in ROS production than Prex1, implying that norbin also has other effectors.
Norbin deficiency increases neutrophil degranulation, phagocytosis, and NET production
In addition to ROS, neutrophils use degranulation, phagocytosis, and NETs to kill bacteria. Degranulation is a process whereby lytic granule contents are secreted and receptors stored on granules upregulated to the plasma membrane. We measured the constitutive and fMLP-stimulated degranulation of gelatinase using TNF-α/GM-CSF priming and cytochalasin B to induce maximal release. NcdnΔmye neutrophils secreted more gelatinase activity than Ncdnfl/fl cells upon incubation at 37°C, which was overcome by increasing doses of fMLP, priming, and cytochalasin B (Figure 5A). Therefore, norbin-deficient neutrophils exhibit constitutive degranulation, although stimulated degranulation and overall granule content are normal. Because degranulation raises receptor surface levels, this might explain in part the enhanced neutrophil responsiveness. Phagocytosis of zymosan particles was also increased in NcdnΔmye compared with Ncdnfl/fl and LysMCre neutrophils (Figure 5B). Similarly, the S aureus–induced production of NETs was increased in NcdnΔmye cells compared with Ncdnfl/fl cells, without constitutive NET production (Figure 5C). Norbin deficiency did not cause constitutive production of TNF-α, CXCL1, or interleukin-6, nor affect their zymosan–induced levels (supplemental Figure 7). Hence, norbin deficiency increases major antibacterial responses of neutrophils without causing neutrophil autoactivation.

Myeloid norbin deficiency increases degranulation, phagocytosis, and NET production. (A) Degranulation. Purified Ncdnfl/fl and NcdnΔmye neutrophils were kept on ice for 45 minutes or were primed with 20 ng/mL TNF-α, 50 ng/mL GM-CSF at 37°C for 45 minutes before stimulation with increasing concentrations of fMLP and 10 μM cytochalasin B (CytB) for 30 minutes at 37°C, as indicated. Gelatinase activity released into the supernatant was analyzed by using in-gel zymography. Left-hand panels indicate representative Coomassie-stained gelatin gels, showing digestion of the gel by gelatinase as white areas. The right-hand panel shows the quantification of gelatinase activity by densitometry, expressed as percentage of primed cells. Data are mean ± standard error of the mean of 7 independent experiments; each dot represents 1 experiment. Statistics comprised two-way analysis of variance with Šidák’s multiple comparisons test. (B) Phagocytosis. Ncdnfl/fl, LysMCre, and NcdnΔmye neutrophils were primed with 20 ng/mL TNF-α, 50 ng/mL GM-CSF for 45 minutes at 37°C, plated onto glass coverslips for 30 minutes, and stimulated with zymosan (5 particles/neutrophil) for 30 minutes at 37°C, fixed, stained with FITC-Gr1 antibody and 4′,6-diamidino-2-phenylindole (DAPI), and assessed for the number of zymosan particles within neutrophils by using widefield microscopy and ImageJ analysis. Left-hand panels show representative images of Ncdnfl/fl cells. The right-hand panel shows the quantification by ImageJ analysis. Data are mean ± standard error of the mean of 3 independent experiments; each dot represents 1 experiment. Statistics comprised one-way analysis of variance with Tukey’s multiple comparisons test. (C) NET production. Purified Ncdnfl/fl and NcdnΔmye neutrophils were seeded onto glass slides and allowed to adhere for 30 minutes at 37°C before being stimulated with serum-opsonized S aureus (10 bacteria per neutrophil; filled symbols), or mock-stimulated (open symbols), for the indicated periods of time. Sytox Green dye was added to samples 15 minutes before their time point, samples were live imaged, and NETs quantified by using ImageJ software. Phase contrast was used to count total cells. Left-hand panels show representative images. The red arrow denotes a dead cell, the white arrow a NET. The right-hand panel shows the quantification according to ImageJ analysis. Data are mean ± standard error of the mean of 3 independent experiments. Statistics comprised two-way analysis of variance with Šidák’s multiple comparisons test.
Myeloid norbin deficiency increases degranulation, phagocytosis, and NET production. (A) Degranulation. Purified Ncdnfl/fl and NcdnΔmye neutrophils were kept on ice for 45 minutes or were primed with 20 ng/mL TNF-α, 50 ng/mL GM-CSF at 37°C for 45 minutes before stimulation with increasing concentrations of fMLP and 10 μM cytochalasin B (CytB) for 30 minutes at 37°C, as indicated. Gelatinase activity released into the supernatant was analyzed by using in-gel zymography. Left-hand panels indicate representative Coomassie-stained gelatin gels, showing digestion of the gel by gelatinase as white areas. The right-hand panel shows the quantification of gelatinase activity by densitometry, expressed as percentage of primed cells. Data are mean ± standard error of the mean of 7 independent experiments; each dot represents 1 experiment. Statistics comprised two-way analysis of variance with Šidák’s multiple comparisons test. (B) Phagocytosis. Ncdnfl/fl, LysMCre, and NcdnΔmye neutrophils were primed with 20 ng/mL TNF-α, 50 ng/mL GM-CSF for 45 minutes at 37°C, plated onto glass coverslips for 30 minutes, and stimulated with zymosan (5 particles/neutrophil) for 30 minutes at 37°C, fixed, stained with FITC-Gr1 antibody and 4′,6-diamidino-2-phenylindole (DAPI), and assessed for the number of zymosan particles within neutrophils by using widefield microscopy and ImageJ analysis. Left-hand panels show representative images of Ncdnfl/fl cells. The right-hand panel shows the quantification by ImageJ analysis. Data are mean ± standard error of the mean of 3 independent experiments; each dot represents 1 experiment. Statistics comprised one-way analysis of variance with Tukey’s multiple comparisons test. (C) NET production. Purified Ncdnfl/fl and NcdnΔmye neutrophils were seeded onto glass slides and allowed to adhere for 30 minutes at 37°C before being stimulated with serum-opsonized S aureus (10 bacteria per neutrophil; filled symbols), or mock-stimulated (open symbols), for the indicated periods of time. Sytox Green dye was added to samples 15 minutes before their time point, samples were live imaged, and NETs quantified by using ImageJ software. Phase contrast was used to count total cells. Left-hand panels show representative images. The red arrow denotes a dead cell, the white arrow a NET. The right-hand panel shows the quantification according to ImageJ analysis. Data are mean ± standard error of the mean of 3 independent experiments. Statistics comprised two-way analysis of variance with Šidák’s multiple comparisons test.
The altered distribution of NcdnΔmye neutrophils in the pneumococcus-infected lung prompted us to assess neutrophil adhesion. NcdnΔmye neutrophils exhibited increased adhesion and spreading under all conditions tested (primed or unprimed, fMLP- or mock-stimulated), whereas polarization was normal (supplemental Figure 8). NcdnΔmyePrex1/2−/− cells also exhibited increased adhesion and a rescue of the spreading defect in Prex1/2−/− cells. Therefore, norbin deficiency increases neutrophil adhesion and spreading, largely independently of Prex1.
Norbin deficiency increases the cell surface levels of some GPCRs but not other receptors or adhesion molecules
The increased responsiveness suggested that receptor levels on the surface of norbin-deficient neutrophils may be elevated. Because norbin regulates GPCR trafficking,20,28 we tested the cell surface levels of selected neutrophil GPCRs. Cells were either kept on ice to maintain basal receptor levels, incubated at 37°C to allow constitutive trafficking, or primed to induce maximal receptor upregulation. The effectiveness of these treatments was verified by monitoring the surface level of β2-integrin CD11b (Mac-1) (supplemental Figure 9). In some instances, cells were stimulated to elicit agonist-induced receptor internalization. In basal cells, surface levels of C5aR1 were low and did not differ between NcdnΔmye and Ncdnfl/fl cells, but TNF-α/GM-CSF priming, or incubation at 37°C, induced significantly higher upregulation in NcdnΔmye cells (Figure 6A). This might explain the increased C5a-stimulated ROS production in NcdnΔmye cells. The agonist-induced interalization of C5aR1 was normal, however, as expected. Surface levels of CXCR4 were also higher in NcdnΔmye cells than in Ncdnfl/fl cells, whereas CXCR1 and CXCR2 were unaffected, as was CXCR2 shedding from the neutrophil surface upon priming42; this was in accordance with the constitutive plasma membrane localization of the latter GPCRs43 and the inability of norbin to bind CXCR1.29 The surface levels of several other types of receptors and adhesion molecules (L-selectin, PSGL-1, and the β2-integrins LFA-1 and Mac-1) were unaffected, despite cells reacting normally to priming by shedding L-selectinand upregulating Mac-1 (Figure 6B). β2-integrin avidity was also largely normal, in terms of the number, size (data not shown), and polarization of β2-integrin clusters; the exception was a reduction in the proportion of NcdnΔmye cells that exhibited β2-integrin clusters upon fMLP stimulation (Figure 6C).

Norbin-deficient neutrophils have increased cell surface levels of the GPCRs C5aR1 and CXCR4 but normal surface levels of other receptors and adhesion molecules. (A) Ncdnfl/fl and NcdnΔmye bone marrow cells were kept on ice (basal; open symbols) or were primed with 20 ng/mL TNF-α, 50 ng/mL GM-CSF for 45 minutes at 37°C before transfer onto ice (filled symbols). Cells were stained to identify neutrophils and quantify the mean levels of C5aR1, CXCR4, CXCR1, and CXCR2 on the neutrophil surface by flow cytometry. (Top, middle panel) Bone marrow cells were incubated for 30 minutes at 37°C without or with 100 nM C5a, or C5a was added for the last 10 minutes as indicated, before transfer onto ice and quantification of the mean C5aR1 level on the neutrophil surface. Data are mean ± standard error of the mean (SEM) of 3 independent experiments; each dot is the mean of 1 experiment. (B) Ncdnfl/fl, LysMCre, and NcdnΔmye bone marrow cells were treated as in panel A and stained to identify neutrophils and quantify the mean levels of L-selectin, PSGL-1, LFA-1, and Mac-1 on the neutrophil surface by flow cytometry. Data are mean ± SEM of 3 to 8 independent experiments; each dot is the mean of 1 experiment. (C) Integrin avidity. Purified Ncdnfl/fl and NcdnΔmye neutrophils were primed with 20 ng/mL TNF-α, 50 ng/mL GM-CSF for 45 minutes at 37°C before incubation with AF594-labeled anti-CD18 antibody for 3 minutes at 37°C in the presence (filled symbols) or absence (open symbols) of 1.5 μM fMLP. Cells were fixed and allowed to settle onto electrostatically charged slides before imaging and image analysis for the presence and localization of integrin clusters. A total of 30 to 40 cells were analyzed/condition/experiment. Data are mean ± SEM of 3 independent experiments; each dot represents 1 experiment. Statistics in panels A to C comprised two-way analysis of variance with Šidák’s multiple comparisons test.
Norbin-deficient neutrophils have increased cell surface levels of the GPCRs C5aR1 and CXCR4 but normal surface levels of other receptors and adhesion molecules. (A) Ncdnfl/fl and NcdnΔmye bone marrow cells were kept on ice (basal; open symbols) or were primed with 20 ng/mL TNF-α, 50 ng/mL GM-CSF for 45 minutes at 37°C before transfer onto ice (filled symbols). Cells were stained to identify neutrophils and quantify the mean levels of C5aR1, CXCR4, CXCR1, and CXCR2 on the neutrophil surface by flow cytometry. (Top, middle panel) Bone marrow cells were incubated for 30 minutes at 37°C without or with 100 nM C5a, or C5a was added for the last 10 minutes as indicated, before transfer onto ice and quantification of the mean C5aR1 level on the neutrophil surface. Data are mean ± standard error of the mean (SEM) of 3 independent experiments; each dot is the mean of 1 experiment. (B) Ncdnfl/fl, LysMCre, and NcdnΔmye bone marrow cells were treated as in panel A and stained to identify neutrophils and quantify the mean levels of L-selectin, PSGL-1, LFA-1, and Mac-1 on the neutrophil surface by flow cytometry. Data are mean ± SEM of 3 to 8 independent experiments; each dot is the mean of 1 experiment. (C) Integrin avidity. Purified Ncdnfl/fl and NcdnΔmye neutrophils were primed with 20 ng/mL TNF-α, 50 ng/mL GM-CSF for 45 minutes at 37°C before incubation with AF594-labeled anti-CD18 antibody for 3 minutes at 37°C in the presence (filled symbols) or absence (open symbols) of 1.5 μM fMLP. Cells were fixed and allowed to settle onto electrostatically charged slides before imaging and image analysis for the presence and localization of integrin clusters. A total of 30 to 40 cells were analyzed/condition/experiment. Data are mean ± SEM of 3 independent experiments; each dot represents 1 experiment. Statistics in panels A to C comprised two-way analysis of variance with Šidák’s multiple comparisons test.
Hence, norbin deficiency leads to the upregulation of some GPCRs on the neutrophil surface without affecting agonist-induced GPCR internalization or the surface levels of several other types of receptors and adhesion molecules.
Norbin deficiency increases GPCR-dependent Rac and Erk signaling
To gain further insight into the underlying mechanisms, we investigated neutrophil GPCR signaling. Because norbin activates P-Rex1,13 and P-Rex1 activates Rac upon GPCR stimulation,6,8,10 we measured Rac activity (GTP loading). fMLP-stimulated Rac1 and Rac2 activity was elevated in NcdnΔmye neutrophils at all concentrations of fMLP tested, whereas basal Rac activity was near the detection limit (Figure 7A). Rac1 activity was increased twofold and Rac2 activity 2.5-fold in NcdnΔmye cells compared with Ncdnfl/fl and LysMCre cells. Rac1 activity was raised similarly in NcdnΔmyePrex1/2−/− cells, whereas Rac2 activity was not (Figure 7B). This shows that norbin-dependent regulation of Rac2 (but not Rac1) requires Prex1, in accordance with the substrate preference of this GEF.10 To search for another Rac-GEF that might be controlled by norbin, we tested Vav, which also mediates neutrophil GPCR signaling and has a substrate preference for Rac1.8,9 Vav is regulated by tyrosine phosphorylation, allowing us to test its activity by phospho-western blotting. Mock-stimulated NcdnΔmye neutrophils had higher Vav activity than NcdnΔmye cells (Figure 7C), whereas fMLP-stimulated Vav activity was not consistently elevated. Therefore, norbin controls constitutive Vav activity, identifying Vav as a norbin effector.

Norbin-deficient neutrophils have increased fMLP-stimulated Rac and Erk activities. (A) fMLP dose response of Rac1 and Rac2 activation. Ncdnfl/fl and NcdnΔmye neutrophils were stimulated with the indicated concentrations of fMLP for 10 seconds, lysed, and active, GTP-loaded Rac1 and Rac2 were isolated by PAK-CRIB pull down. Representative blots shown are from 1 of 3 independent experiments. (B) Quantification of Rac1 and Rac2 activities. Neutrophils of the indicated genotypes were stimulated with 10 μM fMLP for 10 seconds (filled symbols), or were mock-stimulated (open symbols), before PAK-CRIB pull down. Top panel: Representative western blots for Rac1 activity are shown compared with 2% of total cell lysate. Bottom panels: Rac1 and Rac2 activities were quantified by densitometry and normalized to fMLP-stimulated Ncdnfl/fl samples. Data are mean ± standard error of the mean of 3 to 7 independent experiments; each dot is the mean of 1 experiment. Statistical analyses were conducted on log-transformed raw data by two-way analysis of variance with Dunnett’s multiple comparisons test. (C) Vav activity. Purified Ncdnfl/fl and NcdnΔmye neutrophils were primed with 20 ng/mL TNF-α, 50 ng/mL GM-CSF for 45 minutes at 37°C, stimulated with 1 µM fLMP for 10 seconds (filled symbols), or mock-stimulated (open symbols), and total cell lysates underwent western blotting for active Vav and total Vav. Left-hand panel: Representative blots. Right-hand panel: quantification by ImageJ analysis of 6 independent experiments; each dot represents 1 experiment. Statistics comprised two-way analysis of variance with Šidák’s multiple comparisons test. (D) Erk activity. Purified Ncdnfl/fl and Ncdn‒/‒ neutrophils were primed with 20 ng/mL TNF-α, 50 ng/mL GM-CSF for 45 minutes at 37°C, stimulated with the indicated doses of fMLP for 45 seconds, and then subjected to western blotting for active and total Erk1/2. Blots shown are from 1 of 6 independent experiments. Quantification of these and related data are shown in supplemental Figure 10. Effects of the Rac inhibitor EHT 1864 (E) and Erk1/2 inhibitor SCH772984 (F) on ROS production. Neutrophils were incubated with the indicated doses of inhibitor for 1 hour during priming with TNF-α, GM-CSF, before stimulation of ROS production with fMLP as in supplemental Figure 6A. Data are mean AUC ± standard error of the mean from 4 independent experiments. Statistics comprised two-way analysis of variance with Šidák’s multiple comparisons test. Values for 50% inhibitory concentration (IC50) were calculated from nonlinear best-fit curves on data normalized to maximal ROS produced.
Norbin-deficient neutrophils have increased fMLP-stimulated Rac and Erk activities. (A) fMLP dose response of Rac1 and Rac2 activation. Ncdnfl/fl and NcdnΔmye neutrophils were stimulated with the indicated concentrations of fMLP for 10 seconds, lysed, and active, GTP-loaded Rac1 and Rac2 were isolated by PAK-CRIB pull down. Representative blots shown are from 1 of 3 independent experiments. (B) Quantification of Rac1 and Rac2 activities. Neutrophils of the indicated genotypes were stimulated with 10 μM fMLP for 10 seconds (filled symbols), or were mock-stimulated (open symbols), before PAK-CRIB pull down. Top panel: Representative western blots for Rac1 activity are shown compared with 2% of total cell lysate. Bottom panels: Rac1 and Rac2 activities were quantified by densitometry and normalized to fMLP-stimulated Ncdnfl/fl samples. Data are mean ± standard error of the mean of 3 to 7 independent experiments; each dot is the mean of 1 experiment. Statistical analyses were conducted on log-transformed raw data by two-way analysis of variance with Dunnett’s multiple comparisons test. (C) Vav activity. Purified Ncdnfl/fl and NcdnΔmye neutrophils were primed with 20 ng/mL TNF-α, 50 ng/mL GM-CSF for 45 minutes at 37°C, stimulated with 1 µM fLMP for 10 seconds (filled symbols), or mock-stimulated (open symbols), and total cell lysates underwent western blotting for active Vav and total Vav. Left-hand panel: Representative blots. Right-hand panel: quantification by ImageJ analysis of 6 independent experiments; each dot represents 1 experiment. Statistics comprised two-way analysis of variance with Šidák’s multiple comparisons test. (D) Erk activity. Purified Ncdnfl/fl and Ncdn‒/‒ neutrophils were primed with 20 ng/mL TNF-α, 50 ng/mL GM-CSF for 45 minutes at 37°C, stimulated with the indicated doses of fMLP for 45 seconds, and then subjected to western blotting for active and total Erk1/2. Blots shown are from 1 of 6 independent experiments. Quantification of these and related data are shown in supplemental Figure 10. Effects of the Rac inhibitor EHT 1864 (E) and Erk1/2 inhibitor SCH772984 (F) on ROS production. Neutrophils were incubated with the indicated doses of inhibitor for 1 hour during priming with TNF-α, GM-CSF, before stimulation of ROS production with fMLP as in supplemental Figure 6A. Data are mean AUC ± standard error of the mean from 4 independent experiments. Statistics comprised two-way analysis of variance with Šidák’s multiple comparisons test. Values for 50% inhibitory concentration (IC50) were calculated from nonlinear best-fit curves on data normalized to maximal ROS produced.
To analyze other fMLP-stimulated signaling pathways, neutrophil lysates of TNF-α/GM-CSF–primed neutrophils were subjected to western blotting for active Erk, p38Mapk, Jnk, and Akt. At the peak of stimulation (1 μM fMLP, 45 seconds), Erk activity was twofold higher in NcdnΔmye neutrophils than in the Ncdnfl/fl neutrophils. No significant differences were seen in the other signaling pathways under any conditions tested (Figure 7D; supplemental Figure 10). Hence, norbin deficiency increases fMLP-stimulated Erk activity without obviously affecting several other GPCR-signaling pathways, proposing a degree of pathway specificity.
These data suggested a mechanism whereby raised GPCR surface levels and GPCR-dependent Rac and Erk signaling underlie the enhanced responsiveness of norbin-deficient neutrophils. To test this hypothesis, we assessed the contribution of Rac and Erk activities to ROS production using the Rac inhibitor EHT 1864 and Erk1/2 inhibitor SCH772984. Both inhibitors blocked fMLP-stimulated ROS production in NcdnΔmye and Ncdnfl/fl neutrophils with 50% inhibitory concentration values in the low micromolar range that were only marginally (twofold to threefold) higher in NcdnΔmye cells (Figure 7E-F); this finding is consistent with the increased Rac and Erk activities contributing to the elevated ROS production in norbin-deficient cells.
Discussion
We found that myeloid norbin deficiency improves immunity against pneumococcus by increasing the involvement of neutrophils in clearing this infection. It increases neutrophil responsiveness, promoting the ability of neutrophils to kill bacteria in a ROS-dependent manner, enhancing phagocytosis, degranulation, and the production of ROS and NETs. It raises GPCR levels on the neutrophil surface and facilitates GPCR signaling through Rac and Erk.
Pneumococcal infection continues to be a major challenge to human health worldwide. New avenues are being sought to enhance particularly the early innate immune response to this infection, which is afforded by macrophages and neutrophils, the latter mainly when alveolar macrophages are overwhelmed by bacterial titer, comorbidity, coinfection, and immunosuppressive states such as young or old age.2 Mouse models recapitulate the human immune response, showing that neutrophils are more important for clearing pneumococcal infection in neonates than in adults41 and at higher bacterial titers.40,44,45 We tested the early response under conditions that result in 60% of mice dying after 4 days if the disease is allowed to progress.32 Under these conditions, neutrophils were nonessential for clearing pneumococcus in control animals, but norbin-deficient mice cleared an order of magnitude more bacteria than control mice. Depletion of neutrophils or macrophages showed that this increased immunity stemmed from neutrophils, suggesting that norbin deficiency renders neutrophils more important for combating infection. We did not perform lethality curves for ethical and legal reasons, but increased survival of norbin-deficient mice would be expected. Histology, assessment of lung lavages/homogenates, and chemokine/cytokine analysis showed that norbin deficiency does not cause autoinflammation or hyperinflammation in response to infection. Normal numbers of neutrophils were recruited, although neutrophil distribution within the infected lung was altered. The latter suggests that neutrophils had either progressed further in the recruitment process, or they took a different route, which warrants further investigation by intravital microscopy. Norbin-deficient mice also cleared an order of magnitude more bacteria in a model of septic peritonitis, showing that the immune-suppressor role of norbin is not restricted to pneumococcal infection.
Neutrophils use a range of responses to kill bacteria, including degranulation, phagocytosis, and the production of ROS and NETs. We found that all these responses are increased with norbin deficiency, together with a ROS-dependent increase in bacterial killing. Hence, it seems that neutrophils express norbin as a brake mechanism to prevent excessive activation, perhaps so they do not become fully activated before they reach the site of infection, or to increase their ability to coordinate phagocytosis, degranulation, and ROS and NET production into a concerted antibacterial response. Neutrophil responsiveness remains to be assessed in vivo. Neutrophils can mediate much of the tissue damage associated with pneumococcal infection,2 independently of ROS,45 but inflammation markers were normal and neutrophils not autoactivated. However, unlike the acute infection conditions tested here, chronic infectious/inflammatory conditions or old age might reveal detrimental effects of norbin deficiency such as tissue injury or immune dysregulation. It remains to be seen if ROS contribute to the increased immunity, given that gp91phox−/− mice are not impaired in clearing pneumococcus.46 It also remains to be investigated if faster or different migration modes bring about the altered neutrophil distribution within the infected lung, considering the increased neutrophil adhesion but largely normal β2-integrin avidity in vitro.
It is unknown whether norbin is governed by any form of regulation. Norbin has no discernible domains and is predicted to be entirely composed of armadillo repeat α-helices.14 It confers neurite outgrowth through its 100 N-terminal amino acids16 and binds the GPCRs through its C-terminal third.20,28 Intramolecular autoinhibition may exist, as the N-terminus has greater neurite outgrowth-promoting activity than the full-length protein,16 but it is unknown if any signals can relieve this putative autoinhibition. The protein is largely cytosolic, although palmitoylation can target it to endosomes47 and coexpression with P-Rex1 to the plasma membrane.13 It will be interesting to investigate subcellular localization and binding to GPCRs and Prex1 to evaluate mechanisms of norbin regulation in neutrophils.
Norbin regulates the steady-state trafficking of GPCRs that it binds directly,20,28,29 but the primary sequence is insufficient to predict which GPCRs norbin can bind,20,29 and thus this needs to be determined empirically. We report raised cell surface levels of C5aR1 and CXCR4 in norbin-deficient neutrophils, suggesting that these receptors may be norbin targets. Norbin does not control agonist-stimulated GPCR internalization,28 and C5aR1 surface levels were indeed normal upon C5a stimulation. The surface levels of CXCR1, CXCR2, and several other receptors and adhesion molecules were also normal. C5aR1 and CXCR4 are stored on granules and upregulated by degranulation, whereas CXCR1 and CXCR2 are constitutively plasma membrane localized. The increased constitutive degranulation may explain how C5aR1 and CXCR4 become upregulated in norbin-deficient cells, but unlike Mac-1, which was trafficked normally, these receptors also seemed to be preferentially retained at the membrane. Thus, we speculate that norbin controls both the upregulation through degranulation and the recycling of its target GPCRs.
Norbin deficiency increased fMLP-stimulated Rac and Erk activities without obviously affecting several other GPCR-signaling pathways. These elevated Rac and Erk activities might underlie the increased responsiveness of norbin-deficient cells, as suggested by use of Rac and Erk1/2 inhibitors. We previously reported that norbin stimulates P-Rex1 Rac-GEF activity, concluding that norbin is a positive regulator of P-Rex1.13 This conclusion stands, but our current study shows that norbin has more fundamental, largely Prex1-independent roles in neutrophils, which implied that norbin has additional effectors. We identified the Rac-GEF Vav, which activates Rac1 in neutrophil GPCR signaling,8,9,48 as one such norbin effector. Norbin-mediated regulation of Erk was previously described but is context dependent and the mechanism unknown.20,28 We speculate that norbin may control Erk through Ras and Ras-GEFs, in a similar manner to its regulation of Rac and Rac-GEFs. An unbiased approach will be required for a comprehensive understanding of neutrophil receptors and signaling pathways controlled by norbin.
In summary, we showed that myeloid norbin deficiency increases neutrophil responsiveness and neutrophil-mediated antibacterial immunity. Future research should investigate the full scope of myeloid norbin as an immune-suppressor, the underlying molecular mechanisms, and the potential of norbin as a target in immune diseases.
This project was funded by Institute Strategic Programme Grant BB/P013384/1 from the Biotechnology and Biological Sciences Research Council (BBSRC) to the Signalling Programme at the Babraham Institute. C.P. was, and P.M. currently is, the recipient of a targeted studentship from the BBSRC, and S.C. is the recipient of a targeted studentship from the Medical Research Council. E.H. receives an iCASE studentship from the BBSRC.
The authors dedicate this article to the memory of Michael Wakelam, the director of their institute, who died of complications of a suspected COVID-19 infection.
Acknowledgments
The authors thank Sonja Vermeren, Centre for Inflammation Research, for her kind advice on conditions for the degranulation assay. They also thank the staff of the Babraham Biological Support Unit and the Imaging and Flow Cytometry facilities for their dedicated and highly skilled support.
Authorship
Contribution: C.P. designed research, collected and analyzed data, and wrote the paper; D.P., S.C., A.-K.S., K.H., P.M., and M.J.B. designed research, and collected and analyzed data; L.C., E.H., and A.M. collected and analyzed data; R.W. and A.S.-P. designed the analytical plan, designed research, and analyzed data; Y.R.V., C.V.V., S.J.C and S.P. designed research; K.O. critically revised the manuscript for important intellectual content; and H.C.E.W. designed the analytical plan, designed research, collected and analyzed data, wrote the paper, and critically revised it for important intellectual content.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Heidi C.E. Welch, Signalling Programme, The Babraham Institute, Babraham Research Campus, Cambridge CB22 3AT, United Kingdom; e-mail: heidi.welch@babraham.ac.uk.

