

TO THE EDITOR:
Porphyria is a group of genetic and acquired disorders caused by the accumulation of porphyrins or porphyrin precursors because of defects in the heme biosynthetic pathway.1 Depending on the source of accumulated porphyrins or their precursors and duration of the porphyric attacks, porphyrias are often classified as acute hepatic porphyrias, chronic hepatic porphyrias, or erythropoietic porphyrias.2 Clinical manifestations of porphyrias may include skin photosensitivity, neurovisceral symptoms and seizures, liver damage, and other organ involvement.3 Although porphyria had been studied for more than a century, the exact biochemical mechanism of porphyrin-induced tissue damage is not clear. Several lines of evidence3-9 suggest a unique mechanism for porphyrin-induced tissue damage that involves porphyrin-mediated protein oxidation and aggregation. As part of this mechanism, porphyrin binding causes localized unfolding10 and conformational changes in native protein structures, with subsequent photosensitization of the bound porphyrin molecule leading to targeted oxidation of select methionine or other targeted residues.3,7 Noncovalent (eg, hydrophobic, π-π ionic interactions) porphyrin-porphyrin and porphyrin-oxidized proteins interactions cause the proteins to aggregate.3 In acute porphyrias that involve increased δ-aminolevulinic acid (ALA)/porphobilinogen (PBG), protein aggregation has not been described; we posit that ALA/PBG cross the blood–brain barrier11 and then enter neurons with potential porphyrin accumulation and protein aggregation.3 Porphyrin-mediated protein aggregation and subsequent loss of function can have debilitating consequences in cellular homeostasis. Organelle-specific and targeted protein aggregation by fluorescent porphyrins is a likely major mechanism of tissue damage in porphyria.3,7
Among the 8 different genetic porphyrias, most published reports place porphyria cutanea tarda (PCT) as the most common porphyria in the world, (Figure 1; supplemental Table 1). PCT is caused by a deficiency in uroporphyrinogen decarboxylase (UROD), that leads to accumulation of colorless uroporphyrinogen,12 which, in turn, is oxidized to uroporphyrin by CYP1A2,13 spontaneously,14 or due to other factors.15 There are 2 major types of PCT, type I (acquired), and type II (familial). In type II PCT, an inherited autosomal dominant UROD mutation leads to decrease of UROD activity. The more common type I PCT is caused by iron overload coupled with several associated susceptibility factors including smoking, alcoholism, hemochromatosis (HFE), or estrogen use.16-18 Iron overload promotes uroporphyrinogen oxidation to uroporphomethene, a competitive inhibitor of UROD,19 and subsequent acquired PCT.16,17 Both type I and type II PCT require the presence of susceptibility factors.16 The second most common porphyria is acute intermittent porphyria (AIP), caused by mutation in hydroxymethylbilane synthase (HMBS), that leads to accumulation of ALA and PBG.1 The prevalence of disease-causing mutations in HMBS among Whites is 600 to 700 cases/million.20 However, the clinical penetrance of AIP (Figure 1; supplemental Tables 1 and 5) is only 1% to 2%, which underscores the effect of additional genetic, environmental, and host factors. For example, AIP is mainly a clinical disease that involves women of reproductive age in 80% to 90% of cases.21 This striking effect of secondary modifiers is much more apparent when we examine the estimated reported prevalence of PCT and AIP in mainland China (Figure 1; supplemental Table 1).
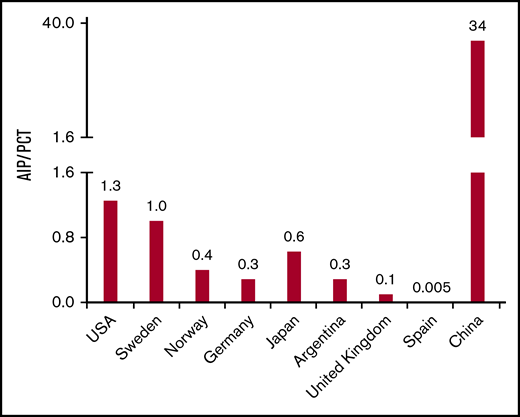
Frequency ratio of AIP to PCT among different geographic regions. For all listed regions, except China, the frequency ratio of AIP to PCT is obtained by dividing the prevalence rates of AIP to PCT in the indicated region (supplemental Table 1). For China, the frequency ratio of AIP to PCT is estimated by dividing the number of AIP cases by the number of PCT cases as reported between 2003 and 2019 in the Chinese medical database Wanfang Med Online (WMO; http://med.wanfangdata.com.cn/).
Frequency ratio of AIP to PCT among different geographic regions. For all listed regions, except China, the frequency ratio of AIP to PCT is obtained by dividing the prevalence rates of AIP to PCT in the indicated region (supplemental Table 1). For China, the frequency ratio of AIP to PCT is estimated by dividing the number of AIP cases by the number of PCT cases as reported between 2003 and 2019 in the Chinese medical database Wanfang Med Online (WMO; http://med.wanfangdata.com.cn/).
Herein, we searched WMO (http://eng.med.wanfangdata.com.cn/), the major repository of clinical and biomedical articles in China. WMO is a medical branch of Wanfang Data, and it indexes more than 220 Chinese medical journals that are published mostly in Mandarin. WMO is the sole source for physicians to access full electronic versions of the top 115 medical journals of the Chinese Medical Association. We searched the WMO for the term “porphyria,” with selection of all case reports and manuscripts published from 2003 to 2019. After the 2003 Severe Acute Respiratory Syndrome outbreak, genetic analysis technology was widely introduced in major Chinese hospitals. Because of the lack of genetic testing, porphyria cases before 2003 were mostly unclassified; therefore, for the purpose of this analysis, we only included papers published beginning in 2003. Notably, analysis of porphyria case reports during this 16-year period showed that the pattern of AIP vs PCT prevalence among the Chinese population (AIP>>PCT) is dramatically different from other regions of the world (Figure 1; supplemental Table 1). There were approximately 790 reported patients with AIP (care was taken to exclude potential overlap among the different studies), and 70% were females, which is similar to the sex distribution of AIP cases observed in other countries. The clinical presentations were also similar to what is expected for AIP, including liver injury, peripheral neuropathy, and 10% of patients reporting myalgia and rhabdomyolysis (supplemental Table 2). Several AIP cases described in supplemental Table 2 also displayed novel genetic modifications not described in gnomAD and National Center for Biotechnology Information databases (Table 1). In contrast, only 23 patients with PCT were reported between 2003 and 2019. All the PCT cases were small case studies, whereas 17% (12 of 71) of the AIP studies were larger case series involving 10 or more patients. More than 50% of the PCT patients were exposed to risk factors such as alcohol, hexachlorobenzene, and fertilizer (supplemental Table 3). Notably, the contrasting prevalence trend that was observed among Chinese patients with AIP and PCT, compared with the rest of the world, were not seen in Chinese patients with erythropoietic protoporphyria (EPP; supplemental Table 4). As observed in other regions of the world, EPP cases were also rare (Figure 1; supplemental Table 1), with only 22 reported cases (supplemental Table 4). Notably, the AIP>>PCT trend noted in the Chinese population was not present in other Asian countries such as Japan (Figure 1). This variation in porphyria prevalence predicts likely genetic modifiers that remain to be defined. Several risk factors for PCT, including chronic hepatitis C, excess alcohol intake, and exposure to chemicals such as polychlorinated benzene, are present in the Chinese population. However, the prevalence of iron overload is low, because of the rarity of HFE mutations among Han Chinese.22 This might explain the relatively rare occurrence of PCT in China compared with Europe and highlights the importance of secondary modifiers (genetic, chemical, environmental) in modulating the severity or susceptibly to clinically evident porphyria.
Novel HMBS mutants that cause AIP, and putative genetic modifiers of porphyria
| Novel HMBS mutants | Putative genetic modifiers of porphyria | ||
|---|---|---|---|
| Mutation type | Variant | Classification | Name |
| Deletion | c.6delA | Serum porphyrin transporters | Albumin |
| c.657_658delCT | Haptoglobin | ||
| Hemopexin | |||
| High/low-density lipoproteins | |||
| Insertion | c.741-748dupCATCGCTG | Porphyrin transporters | FLVCR1a/b |
| BCRP/ABCG2 | |||
| ABCB6 | |||
| Substitution | c.93C>T | Cellular porphyrin binding proteins | Glutathione S-transferase |
| c.763G>T | Liver fatty acid binding protein | ||
| c.4219T>G | Heme binding protein 23 | ||
| c.5387C>T | p22HBP | ||
| p.H305N (c.1070C>A) | GAPDH | ||
| p.R321L (c.1119G>T, c.1120T>A) | |||
| Splicing | c.651+2A>G | Porphyrin biosynthesis modulators | CLPX |
| IVS3-1G>C | β-catenin | ||
| GATA-1 | |||
| Novel HMBS mutants | Putative genetic modifiers of porphyria | ||
|---|---|---|---|
| Mutation type | Variant | Classification | Name |
| Deletion | c.6delA | Serum porphyrin transporters | Albumin |
| c.657_658delCT | Haptoglobin | ||
| Hemopexin | |||
| High/low-density lipoproteins | |||
| Insertion | c.741-748dupCATCGCTG | Porphyrin transporters | FLVCR1a/b |
| BCRP/ABCG2 | |||
| ABCB6 | |||
| Substitution | c.93C>T | Cellular porphyrin binding proteins | Glutathione S-transferase |
| c.763G>T | Liver fatty acid binding protein | ||
| c.4219T>G | Heme binding protein 23 | ||
| c.5387C>T | p22HBP | ||
| p.H305N (c.1070C>A) | GAPDH | ||
| p.R321L (c.1119G>T, c.1120T>A) | |||
| Splicing | c.651+2A>G | Porphyrin biosynthesis modulators | CLPX |
| IVS3-1G>C | β-catenin | ||
| GATA-1 | |||
Novel HMBS mutants as identified by comparing the AIP mutants described in supplemental Table 2 with the data for HMBS in the gnomAD database and National Center for Biotechnology Information database. The listed variants are included as reported in the citations in supplemental Table 2. Putative reported genetic modifiers of porphyria, based on the ability of these proteins to bind porphyrins or modulate porphyrin biosynthesis directly or indirectly. For details and primary citations, see reference 3.
ABCB6, adenosine triphosphate-binding cassette subfamily B member 6; ABCG2, adenosine triphosphate-binding cassette subfamily G member 2; BCRP, breast cancer resistance protein; CLPX, adenosine triphosphate-dependent Clp protease adenosine triphosphate-binding subunit clpX-like; FLVCR1a/b, feline leukemia virus subgroup C receptor-related protein 1a/b; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; GATA-1, GATA-binding factor 1; p22HBP, 22-kDa heme-binding protein.
Indeed, several secondary genetic modifiers for porphyria have been predicted (Table 1). For example, mice expressing mutant ABCB6 (porphyrin transporter) accumulated more porphyrin and displayed worse porphyria symptoms compared with porphyric mice with wild-type ABCB6.23 Similarly, patients with severe porphyria carry the same dysfunctional ABCB6 variant that increased porphyrin accumulation in mice.23 Furthermore, hepatic β-catenin modulates experimentally induced porphyria in mice.8 For example, mice lacking β-catenin that were fed a diet containing the porphyrinogenic drug 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) accumulated less hepatic porphyrin and had decreased hepatic protein aggregation and damage compared with wild-type mice fed DDC.8
The data mining from WMO carried out herein has potential limitations that include underreporting, limited genetic/biochemical testing, and different time interval sampling when comparing Chinese with other populations. Although the reported EPP cases in China are rare (population frequency of severe/null FECH mutations is unknown), Chinese and other Asian populations show much higher FECH IVS3-48C polymorphism frequency than Western populations.24 However, homozygous FECH IVS3-48C polymorphism appears to associate with mild EPP,25 which would likely not have been included in the WMO database. Also, potential database underreporting is likely to be similar among the different types of porphyria. We hypothesize that, perhaps, genetic modifiers other than HFE mutations occur in the Chinese population that protect from PCT but predispose to AIP. We also highlight the usefulness of the WMO database, which contains a wealth of clinical information published primarily in Mandarin that has largely remained inaccessible to non–Mandarin-speaking professionals.
For data, e-mail the corresponding author at bishr.omary@rutgers.edu.
Acknowledgment
This work is supported by National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grant R01 DK116548 (M.B.O.).
Contribution: P.L. and M.B.O. designed the analysis; P.L. and N.K. collected data; P.L., N.K., D.M., H.L.B., and M.B.O. analyzed and interpreted data; D.M. and P.L. wrote the manuscript; and all other authors critically read and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Bishr Omary, Center for Advanced Biotechnology and Medicine, 679 Hoes Lane W, Piscataway, NJ 08854; e-mail: bishr.omary@rutgers.edu.

