

Key Points
Traditional (kidney disease) and SCD-specific (SS/Sβ0-thal, HU, low Hb F%, high WBC count, and Hb) risk factors are associated with thrombosis.
Stroke recurrence was 24% despite being at transfusion goal and VTE recurrence was lower with direct oral anticoagulant vs other therapies.

Abstract
Sickle cell disease (SCD) patients are at a four- to 100-fold increased risk for thrombosis compared with the general population, although the mechanisms and risk factors are not clear. We investigated the incidence and predictors for thrombosis in a retrospective, longitudinal cohort of 1193 pediatric and adult SCD patients treated at our institution between January 2008 and December 2017. SCD diagnosis and thrombotic complications were identified using International Classification of Diseases coding and verified through medical chart review. Clinical and laboratory data were extracted from the medical records. With a median follow-up of 6.4 years, 208 (17.4%) SCD patients experienced 352 thrombotic events (64 strokes, 288 venous thromboembolisms [VTE]). Risk factors for stroke included older age and HbSS/Sβ0-genotype and a lower hemoglobin (Hb) F% in the subset of HbSS/Sβ0-genotype patients (P < .05). VTE risk was independently associated with lower estimated glomerular filtration rate, hydroxyurea (HU) use, HbSS/Sβ0 genotype, and higher white blood cell (WBC) counts and Hb (P ≤ .03). Two thrombomodulin gene variants previously associated with thrombosis in the general African American population, THBD rs2567617 (minor allele frequency [MAF] 0.25; odds ratio [OR], 1.5; P = .049) and THBD rs1998081 (MAF, 0.24; OR, 1.5; P = .059), were associated with thrombosis in this cohort. In summary, thrombotic complications are common, and several traditional and SCD-specific risk factors are associated with thrombotic risk. Future studies integrating clinical, laboratory, and genetic risk factors may improve our understanding of thrombosis and guide intervention practices in SCD.
Introduction
Sickle cell disease (SCD) is an inherited red blood cell disorder caused by homozygous or compound heterozygous inheritance of mutations in the β-globin gene that affects 1 in 365 African Americans.1 Vasculopathy is a hallmark of SCD that contributes to the protean acute and chronic complications.2 Thrombosis has been increasingly recognized as a complication of SCD. The hypercoagulable state may be due to increased exposure of phosphatidylserine on the outer surface of red blood cell membranes, increased tissue factor expression on endothelial cells and in circulation, depletion of protein C and S, endothelial dysfunction, and chronic platelet activation.3
Up to 30% of patients will have a cumulative lifetime risk of a clinically overt cerebrovascular accident (CVA), which is the most common arterial thrombotic event in SCD.4 Risk factors for CVA include the hemoglobin (Hb) SS genotype, an elevated white blood cell count, and low Hb concentrations and Hb F%.4 Chronic red blood cell transfusion therapy reduces the risk for subsequent CVA, but 18% of SCD children will have another overt CVA despite achieving transfusion goals.5 Rates of CVA recurrence in SCD adults treated with transfusion therapy are less clear.
The reported cumulative risk for venous thromboembolism (VTE) in SCD ranges from 2.9% in children up to 25.0% in adults.6-9 Although clinical and laboratory risk factors for VTE are not clear, its clinical importance in SCD is highlighted by a 2.3- to 2.9-fold increased risk of death in those with vs without a VTE event.7,9 The genetic basis for VTE risk in SCD has not been previously reported, but variants implicated in other non-SCD African American cohorts may be relevant.10 It is also important to understand how the type and duration of anticoagulant therapy affect the rate of VTE recurrence, which is also unclear.
In a cohort of 1193 SCD patients, we conducted a retrospective, longitudinal study to identify the (1) incidence, (2) clinical and laboratory risk factors, (3) association of genetic variants implicated in African American thrombosis risk, and (4) rates of recurrence based on type and duration of therapy for thrombotic events over a 10-year period.
Methods
We analyzed SCD patients receiving medical care at the University of Illinois at Chicago (UIC) between January 2008 and December 2017. The protocol was approved by the UIC Institutional Review Board prior to extracting the data from the Cerner PowerChart electronic health records (EHRs).
Through a semiautomated algorithm using the UIC hospital enterprise data warehouse, the SCD cohort was identified through encounters with relevant 9th and 10th Clinical Modifications editions of the International Classification of Disease (ICD-9-CM and ICD-10-CM) diagnostic codes for SCD, Hb fractionations, and having either a minimum of 2 outpatient or 1 inpatient encounter between 1 January 2008 and 31 December 2017 (supplemental Table 1).11 SCD genotype was determined by assessing the categorization of ICD coding and Hb fractionation results with manual confirmation by EHR review for those patients that could not be automatically classified. SCD patients were categorized into 4 distinct subgroups: Hb SS disease or Hb Sβ0-thalassemia, Hb Sβ+-thalassemia, Hb SC, or other sickle gene variant.
Baseline demographic and risk factors were queried from our EHR database, including sex, race/ethnicity, past medical history, orthopedic and central venous catheterization procedure data, laboratory values, and medications prescribed. Baseline laboratory, blood pressure, and anthropometric results were determined for each patient using values from outpatient visits. The estimated glomerular filtration rate (eGFR) was calculated using the Chronic Kidney Disease-Epidemiology formula.12 Hydroxyurea use was defined as a logical variable (ever vs never) based on if the patient was prescribed the medication during the study period. Pregnancy data were initially queried using relevant ICD-9/10-CM for pregnancy outcomes (including deliveries and abortions), and then manually confirmed by EHR review to determine number of pregnancy episodes. Orthopedic procedures and central venous catheterizations were queried using pertinent 9th and 10th Procedure Coding system editions of the International Classification of Disease (ICD-9-PCS and ICD-10-PCS). Thromboembolic event diagnoses were queried with pertinent ICD-9/10-CM coding and manually confirmed upon review of diagnostic imaging and/or medical records. Each event was characterized to determine the type of therapy initiated and the duration of anticoagulation. A recurrent thrombotic event was defined as having a second event, confirmed by manual review of the imaging and/or records, during the study time period. The ICD-9 and ICD-10 codes used for identifying pregnancy, procedures, and thrombotic events are provided in the supplemental Table 1.
Genetic risk variants
Genotyping was carried out using Affymetrix Axiom genome-wide Pan-African GeneChip array at the Core Genomics Facility at UIC in DNA isolated from peripheral blood mononuclear cells in 327 SCD patients, as previously described.13 All samples had genotype call rate >95%. Single-nucleotide polymorphisms (SNPs) deviating from Hardy-Weinberg equilibrium (P < .0001) or with a minor allele frequency (MAF) <0.01 were removed.
Candidate SNPs associated with thrombotic risk in the general African American population were tested.10 Allele dosages were associated with thrombotic risk using an additive model, adjusting for the following covariates: age, sex, SCD genotype, hydroxyurea use, and population structure.
Statistical analysis
For univariate analyses, the associations of linear and categorical variables with any thrombotic event, CVA, or VTE were assessed using the Kruskal-Wallis test and Pearson’s χ2 test, respectively. Variables with P < .002 were considered statistically significant after the Bonferroni correction. An initial multivariate Cox proportional hazards model was fit by including covariates with P ≤ .05 from the univariate analyses. Patients who did not experience a thrombosis were censored at the date of last known contact. The final multivariate model was determined by stepwise variable selection, allowing for both backward and forward selection by sequentially selecting models that minimize the Akaike information criterion, adjusting for the following covariates: age, sex, sickle cell genotype, and hydroxyurea use.14 The analyses were conducted using time updated variables for body mass index, systolic blood pressure, white blood cell count, Hb concentration, reticulocyte count, aspartate aminotransferase (AST), and eGFR.15,16 The eGFR, white blood cell count, AST, and baseline Hb F% were log-transformed to achieve a normal distribution. We conducted the following subset analyses for the multivariate models: (1) risk factors for any thrombosis, CVA, and VTE in patients with the Hb SS/Sβ0-thalassemia genotype; (2) risk factors for venous thrombosis in non–catheter-related VTE events. Patients who received an unknown therapy for CVA (n = 1) or VTE (n = 3) events were excluded from the analyses for recurrence based on initial therapy received. Data preprocessing and analysis were accomplished using custom-written scripts in R17 using premade analytical packages, including tidyverse, lubridate, broom, survival, survminer, coin, and tidyforest.18-24 Median values with interquartile range (IQR) are provided.
Results
An initial ICD query identified 1350 potential SCD subjects. We were able to confirm the diagnosis of SCD in 1193 patients by review of Hb fractionations, transfusion history, and medical records. The median age of the cohort was 24 years (IQR, 17-35 years); 57% were female, 55% were treated with hydroxyurea, and 70% were Hb SS/Sβ0-thalassemia genotype (Table 1). Based on ICD procedure coding, there were 83 orthopedic procedures in 52 patients, 1509 central venous catheterizations in 394 patients, and 209 pregnancies in 138 patients between January 2008 and December 2017.
Baseline clinical and laboratory variables in 1193 SCD patients
| Variable | Value |
|---|---|
| Age at first encounter, median (IQR), y | 24 (17-35) |
| Sex, male/female, n (%) | 515 (43.2)/678 (56.8) |
| Race, African American, n (%) | 1123 (94.1) |
| Sickle cell genotype, n (%) | |
| HbSS/SB0-thalassemia | 831 (69.7) |
| HbSC | 290 (24.3) |
| HbSB+-thalassemia | 69 (5.8) |
| Other | 3 (0.3) |
| Hydroxyurea by sickle cell genotype, n/N (%) | |
| Total | 659/1193 (55.2) |
| HbSS/SB0- thalassemia | 560/831 (67.4) |
| HbSC | 77/290 (26.6) |
| HbSB+-thalassemia | 22/69 (31.9) |
| Other | 0/3 (0) |
| Variable | Value |
|---|---|
| Age at first encounter, median (IQR), y | 24 (17-35) |
| Sex, male/female, n (%) | 515 (43.2)/678 (56.8) |
| Race, African American, n (%) | 1123 (94.1) |
| Sickle cell genotype, n (%) | |
| HbSS/SB0-thalassemia | 831 (69.7) |
| HbSC | 290 (24.3) |
| HbSB+-thalassemia | 69 (5.8) |
| Other | 3 (0.3) |
| Hydroxyurea by sickle cell genotype, n/N (%) | |
| Total | 659/1193 (55.2) |
| HbSS/SB0- thalassemia | 560/831 (67.4) |
| HbSC | 77/290 (26.6) |
| HbSB+-thalassemia | 22/69 (31.9) |
| Other | 0/3 (0) |
All thrombotic episodes
With a median follow-up of 6.4 years (IQR, 2.7-9.8 years), 208 (17.4%) SCD patients experienced 352 thrombotic events (CVA: n = 64, VTE: n = 288). The median age at initial thrombosis was 30 years (IQR, 24-43 years) and median time to initial thrombosis from the first encounter was 3.2 years (IQR, 0.8-5.9 years). The distribution for the type of thrombotic event is provided in Figure 1. Differences in variables by thrombosis status are provided in Table 2. Independent risk factors for a thrombotic event on Cox regression analysis included a lower eGFR (P < .001), hydroxyurea use (P = .02), and Hb SS/Sβ0-thalassemia genotype (P = .05), while trends for an association with tobacco use and higher AST were observed (Figure 2). In the subset of Hb SS/Sβ0-thalassemia patients (n = 831), independent risk factors for thrombosis were lower eGFR, higher AST, and hydroxyurea use (P ≤ .02) (supplemental Figure 1).
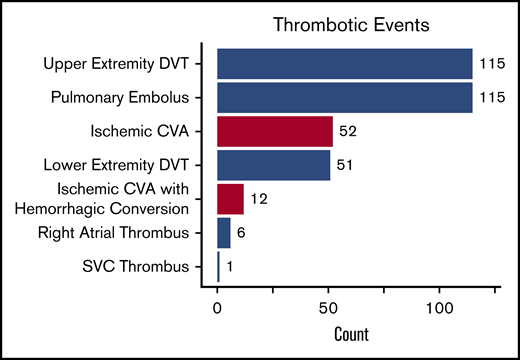
Types of thrombotic events in 1193 SCD patients between January 2008 and December 2017. DVT, deep vein thrombosis; SVC, superior vena cava.
Types of thrombotic events in 1193 SCD patients between January 2008 and December 2017. DVT, deep vein thrombosis; SVC, superior vena cava.
Clinical and laboratory values stratified by thrombotic events in patients with SCD
| n | No thrombosis | n | Thrombosis | P | |
|---|---|---|---|---|---|
| Age at first encounter, y | 985 | 23 (15-35) | 208 | 27 (21-39) | <.0001 |
| Male/female, % | 985 | 45/55 | 208 | 36/64 | .028 |
| African American, n (%) | 985 | 923 (94) | 208 | 200 (96) | .2 |
| Sickle cell genotype, % | 985 | 208 | <.0001 | ||
| HbSS/SB0 thalassemia | 66.7 | 83.7 | |||
| HbSC, SB+-thalassemia, or other | 33.3 | 16.3 | |||
| Hydroxyurea use, % | 985 | 496 (50) | 208 | 163 (78) | <.0001 |
| Tobacco use, % | 985 | 266 (27) | 208 | 87 (42%) | <.0001 |
| Body mass index, kg/m2 | 985 | 22 (18-26) | 208 | 23 (20-27) | .0031 |
| Systolic blood pressure, mm Hg | 985 | 115 (104-127) | 208 | 122 (110-132) | <.0001 |
| White blood cell count, ×103/μL | 985 | 9.7 (7.4-12.2) | 208 | 10.1 (7.6-12.5) | .13 |
| Platelet count, ×103/μL | 985 | 406.0 (287.0-525.0) | 208 | 402.5 (312.5-532.2) | .4 |
| Hb, g/dL | 985 | 9.6 (8.3-11.1) | 208 | 9.0 (8.2-10.2) | .002 |
| Hb F, % | 985 | 4.9 (2.1-11.2) | 208 | 4.7 (2.0-9.2) | .12 |
| Reticulocyte count, ×103/μL | 985 | 249 (158-384) | 208 | 291 (204-423) | .00012 |
| Lactate dehydrogenase, U/L | 985 | 289 (222-408) | 208 | 342 (247-443) | .018 |
| AST, U/L | 985 | 35 (25-47) | 208 | 38 (28-54) | .0016 |
| Serum creatinine, mg/dL | 985 | 0.7 (0.5-0.8) | 208 | 0.7 (0.6-0.9) | <.0001 |
| eGFR, mL/min/1.73 m2 | 985 | 146 (119-171) | 208 | 137 (101-154) | <.0001 |
| Hospitalizations, no. per year | 985 | 1 (0-3) | 208 | 3 (1-7) | <.0001 |
| Pregnancies, n (%) of patients | 545 | 105 (19) | 133 | 33 (25) | .15 |
| Orthopedic procedures, n (%) of patients | 985 | 29 (3) | 208 | 23 (11) | <.0001 |
| Central venous catheters, n (%) of patients | 985 | 244 (25) | 208 | 150 (72) | <.0001 |
| n | No thrombosis | n | Thrombosis | P | |
|---|---|---|---|---|---|
| Age at first encounter, y | 985 | 23 (15-35) | 208 | 27 (21-39) | <.0001 |
| Male/female, % | 985 | 45/55 | 208 | 36/64 | .028 |
| African American, n (%) | 985 | 923 (94) | 208 | 200 (96) | .2 |
| Sickle cell genotype, % | 985 | 208 | <.0001 | ||
| HbSS/SB0 thalassemia | 66.7 | 83.7 | |||
| HbSC, SB+-thalassemia, or other | 33.3 | 16.3 | |||
| Hydroxyurea use, % | 985 | 496 (50) | 208 | 163 (78) | <.0001 |
| Tobacco use, % | 985 | 266 (27) | 208 | 87 (42%) | <.0001 |
| Body mass index, kg/m2 | 985 | 22 (18-26) | 208 | 23 (20-27) | .0031 |
| Systolic blood pressure, mm Hg | 985 | 115 (104-127) | 208 | 122 (110-132) | <.0001 |
| White blood cell count, ×103/μL | 985 | 9.7 (7.4-12.2) | 208 | 10.1 (7.6-12.5) | .13 |
| Platelet count, ×103/μL | 985 | 406.0 (287.0-525.0) | 208 | 402.5 (312.5-532.2) | .4 |
| Hb, g/dL | 985 | 9.6 (8.3-11.1) | 208 | 9.0 (8.2-10.2) | .002 |
| Hb F, % | 985 | 4.9 (2.1-11.2) | 208 | 4.7 (2.0-9.2) | .12 |
| Reticulocyte count, ×103/μL | 985 | 249 (158-384) | 208 | 291 (204-423) | .00012 |
| Lactate dehydrogenase, U/L | 985 | 289 (222-408) | 208 | 342 (247-443) | .018 |
| AST, U/L | 985 | 35 (25-47) | 208 | 38 (28-54) | .0016 |
| Serum creatinine, mg/dL | 985 | 0.7 (0.5-0.8) | 208 | 0.7 (0.6-0.9) | <.0001 |
| eGFR, mL/min/1.73 m2 | 985 | 146 (119-171) | 208 | 137 (101-154) | <.0001 |
| Hospitalizations, no. per year | 985 | 1 (0-3) | 208 | 3 (1-7) | <.0001 |
| Pregnancies, n (%) of patients | 545 | 105 (19) | 133 | 33 (25) | .15 |
| Orthopedic procedures, n (%) of patients | 985 | 29 (3) | 208 | 23 (11) | <.0001 |
| Central venous catheters, n (%) of patients | 985 | 244 (25) | 208 | 150 (72) | <.0001 |
Values are median (IQR) unless otherwise noted.
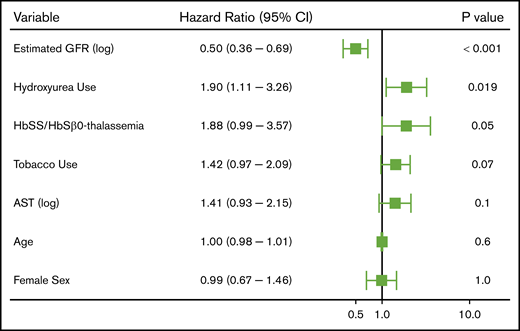
Independent risk factors for a thrombotic event in patients with SCD using a multivariate Cox proportional hazards model. CI, confidence interval.
Independent risk factors for a thrombotic event in patients with SCD using a multivariate Cox proportional hazards model. CI, confidence interval.
CVAs
We observed 64 CVA events in 50 patients (4.2%). The median age at the first CVA event was 30 years (IQR, 20-46 years); 52 (81.3%) were ischemic events and 12 (18.8%) were ischemic events with hemorrhagic conversion. Differences in variables by CVA status are provided in Table 3. Independent risk factors for a CVA event on Cox regression analysis included older age and Hb SS/Sβ0-thalassemia genotype (P = .04) (Figure 3). In the subset of Hb SS/Sβ0-thalassemia genotype patients, a lower Hb F% was an independent risk factor for stroke (P = .046) (supplemental Figure 2).
Clinical and laboratory values stratified by type of thrombotic event in patients with SCD
| n | No thrombosis | n | CVA | n | Venous thrombosis | |
|---|---|---|---|---|---|---|
| Age at first encounter, y | 985 | 23 (15-35) | 50 | 26 (19-40) | 172 | 27 (21-40)** |
| Male/female, % | 985 | 45/55 | 50 | 40/60 | 172 | 35/65* |
| African American, n (%) | 985 | 923 (94) | 50 | 49 (98) | 172 | 165 (96) |
| Sickle cell genotype, n (%) | 985 | 50 | 172 | |||
| HbSS/SB0-thalassemia | 657 (66.7) | 44 (88.0)* | 141 (82.0)** | |||
| HbSC or HbSB+-thalassemia or other | 328 (33.3) | 6 (12.0) | 31 (18.0) | |||
| Hydroxyurea use, n (%) | 985 | 496 (50) | 50 | 34 (68) | 172 | 141 (82)** |
| Tobacco use, n (%) | 985 | 266 (27%) | 50 | 15 (30%) | 172 | 75 (44%)** |
| Body mass index, kg/m2 | 985 | 22 (18-26) | 50 | 23 (20-26) | 172 | 23 (20-28)** |
| Systolic blood pressure, mm Hg | 985 | 115 (104-127) | 50 | 122 (110-130) | 172 | 122 (110-134)** |
| White blood cell count, ×103/μL | 985 | 9.7 (7.4-12.2) | 50 | 9.1 (6.6-12.1) | 172 | 10.2 (7.7-12.9)* |
| Platelet count, ×103/μL | 985 | 406 (287-525) | 50 | 392 (335-569) | 172 | 404 (311-529) |
| Hb, g/dL | 985 | 9.6 (8.3-11.1) | 50 | 8.8 (7.9-9.6)* | 172 | 9.0 (8.3-10.3)* |
| Hb F, % | 985 | 4.9 (2.1-11.2) | 50 | 2.5 (1.1-7.8)* | 172 | 5.2 (2.3-9.9) |
| Reticulocyte count, ×103/μL | 985 | 249 (158-384) | 50 | 283 (249-488)* | 172 | 291 (193-415)* |
| Lactate dehydrogenase, U/L | 985 | 289 (222-408) | 50 | 414 (296-481)** | 172 | 323 (234-440) |
| AST, U/L | 985 | 35 (25-47) | 50 | 44 (30-58)* | 172 | 37 (28-53) |
| Serum creatinine, mg/dL | 985 | 0.7 (0.5-0.8) | 50 | 0.7 (0.6-1.0)* | 172 | 0.7 (0.6-0.9)** |
| eGFR, mL/min/1.73 m2 | 985 | 146 (119-171) | 50 | 138 (95-154)* | 172 | 136 (102-155)** |
| Hospitalizations, no. per year | 985 | 1 (0-3) | 50 | 1 (1-4) | 172 | 4 (1-9)** |
| Pregnancies, n (%) of patients | 545 | 30 | 112 | |||
| Before the first thrombotic event | 105 (19) | 1 (4)* | 23 (21) | |||
| During the entire time period | 3 (10) | 31 (28)* | ||||
| Orthopedic procedures, n (%) of patients | 985 | 50 | 172 | |||
| Before the first thrombotic event | 29 (3) | 3 (6) | 8 (5) | |||
| During the entire time period | 5 (10)* | 19 (11)** | ||||
| Central venous catheters, n (%) of patients | 985 | 50 | 172 | |||
| Before the first thrombotic event | 244 (25) | 16 (32) | 89 (52)** | |||
| During the entire time period | 36 (72)** | 125 (72)** |
| n | No thrombosis | n | CVA | n | Venous thrombosis | |
|---|---|---|---|---|---|---|
| Age at first encounter, y | 985 | 23 (15-35) | 50 | 26 (19-40) | 172 | 27 (21-40)** |
| Male/female, % | 985 | 45/55 | 50 | 40/60 | 172 | 35/65* |
| African American, n (%) | 985 | 923 (94) | 50 | 49 (98) | 172 | 165 (96) |
| Sickle cell genotype, n (%) | 985 | 50 | 172 | |||
| HbSS/SB0-thalassemia | 657 (66.7) | 44 (88.0)* | 141 (82.0)** | |||
| HbSC or HbSB+-thalassemia or other | 328 (33.3) | 6 (12.0) | 31 (18.0) | |||
| Hydroxyurea use, n (%) | 985 | 496 (50) | 50 | 34 (68) | 172 | 141 (82)** |
| Tobacco use, n (%) | 985 | 266 (27%) | 50 | 15 (30%) | 172 | 75 (44%)** |
| Body mass index, kg/m2 | 985 | 22 (18-26) | 50 | 23 (20-26) | 172 | 23 (20-28)** |
| Systolic blood pressure, mm Hg | 985 | 115 (104-127) | 50 | 122 (110-130) | 172 | 122 (110-134)** |
| White blood cell count, ×103/μL | 985 | 9.7 (7.4-12.2) | 50 | 9.1 (6.6-12.1) | 172 | 10.2 (7.7-12.9)* |
| Platelet count, ×103/μL | 985 | 406 (287-525) | 50 | 392 (335-569) | 172 | 404 (311-529) |
| Hb, g/dL | 985 | 9.6 (8.3-11.1) | 50 | 8.8 (7.9-9.6)* | 172 | 9.0 (8.3-10.3)* |
| Hb F, % | 985 | 4.9 (2.1-11.2) | 50 | 2.5 (1.1-7.8)* | 172 | 5.2 (2.3-9.9) |
| Reticulocyte count, ×103/μL | 985 | 249 (158-384) | 50 | 283 (249-488)* | 172 | 291 (193-415)* |
| Lactate dehydrogenase, U/L | 985 | 289 (222-408) | 50 | 414 (296-481)** | 172 | 323 (234-440) |
| AST, U/L | 985 | 35 (25-47) | 50 | 44 (30-58)* | 172 | 37 (28-53) |
| Serum creatinine, mg/dL | 985 | 0.7 (0.5-0.8) | 50 | 0.7 (0.6-1.0)* | 172 | 0.7 (0.6-0.9)** |
| eGFR, mL/min/1.73 m2 | 985 | 146 (119-171) | 50 | 138 (95-154)* | 172 | 136 (102-155)** |
| Hospitalizations, no. per year | 985 | 1 (0-3) | 50 | 1 (1-4) | 172 | 4 (1-9)** |
| Pregnancies, n (%) of patients | 545 | 30 | 112 | |||
| Before the first thrombotic event | 105 (19) | 1 (4)* | 23 (21) | |||
| During the entire time period | 3 (10) | 31 (28)* | ||||
| Orthopedic procedures, n (%) of patients | 985 | 50 | 172 | |||
| Before the first thrombotic event | 29 (3) | 3 (6) | 8 (5) | |||
| During the entire time period | 5 (10)* | 19 (11)** | ||||
| Central venous catheters, n (%) of patients | 985 | 50 | 172 | |||
| Before the first thrombotic event | 244 (25) | 16 (32) | 89 (52)** | |||
| During the entire time period | 36 (72)** | 125 (72)** |
Values are median (IQR) unless otherwise noted.
*P < .05; **P < .002 (statistically significant after Bonferroni correction).
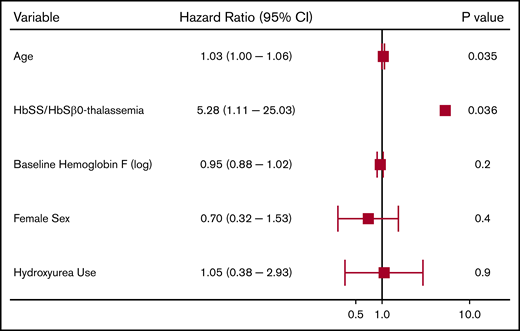
Risk factors for a cerebrovascular event in patients with SCD using a multivariate Cox proportional hazards model.
Risk factors for a cerebrovascular event in patients with SCD using a multivariate Cox proportional hazards model.
We observed 20 recurrent CVA events in 9 (18%) of the 50 SCD patients that experienced an initial CVA. Rates of CVA recurrence did not differ based on initial CVA-related therapy (chronic transfusion, 8/36; anticoagulation, 0/5; aspirin, 0/4; no therapy, 1/4; P = .5). Stroke recurrence was observed in 24% (4/17) of SCD patients with Hb fractionations at goal for chronic transfusion (defined as >80% of Hb fractionations with Hb S ≤30% within the first 2 years and Hb S ≤50% after the first 2 years of a CVA event).5
VTE
We observed 288 VTE events in 172 (14.4%) SCD patients; 194 VTE events occurred in females, and 94 VTE events occurred in males. The median age at the time of the first VTE event was 30 years (IQR, 25-43 years) and included 115 pulmonary embolism, 167 deep vein thrombosis (115 upper extremity, 51 lower extremity, and 1 superior vena cava), and 6 right atrial thrombus events. One-hundred and six (36.8%) of the VTEs occurred within 30 days of a previous hospital discharge. One-hundred and three (35.8%) VTE events were central venous catheter related, while 20 (6.9%) occurred in the perioperative period and 16 (8.2% of VTE events in females) during pregnancy. Differences in variables by VTE status are provided in Table 3. Independent risk factors for VTE risk on Cox regression analysis included lower eGFR (P < .001), hydroxyurea use (P = .003), Hb SS/Sβ0-thalassemia genotype (P = .01), higher Hb concentration (P = .03), and higher white blood cell count (P = .03), while a trend for an association with tobacco use was observed (P = .09) (Figure 4). A lower eGFR, hydroxyurea use, and a higher white blood cell count and Hb concentration were also independent risk factors for VTE in the subset of Hb SS/Sβ0-thalassemia genotype patients (supplemental Figure 3) and in the subset of non–catheter-related VTE events (n = 185) (supplemental Figure 4).
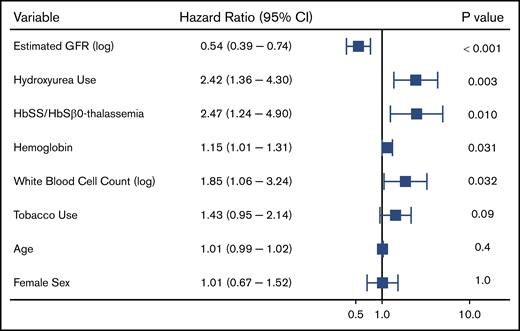
Risk factors for a venous thrombotic event in patients with SCD using a multivariate Cox proportional hazards model.
Risk factors for a venous thrombotic event in patients with SCD using a multivariate Cox proportional hazards model.
Fifty-seven of 172 (33.1%) patients with an initial VTE had recurrent VTE events. VTE recurrence was lower in SCD patients treated with an oral direct factor Xa inhibitor (3/29) compared with other therapies (warfarin, 44/113; low-molecular-weight heparin, 6/16; no therapy, 3/11; P = .008). For those patients started on warfarin, international normalized ratio (INR) results within the designated therapy duration were available for 104 of 113 patients. The proportion of SCD patients on warfarin at goal (defined as >60% of INR results ≥2.0)25 was 17.3% (18/104). We did not observe an association between VTE recurrence and warfarin being at goal (VTE recurrence, 5/41 at goal; no VTE recurrence, 13/63 at goal; P = .4). We did not observe an association between VTE recurrence and duration of anticoagulation therapy (<3 months, 1/6; 3-6 months, 34/97; >6 months, 17/55, P = .8).
African American gene risk variants for thrombosis
Next, we evaluated genetic risk variants that have been implicated in thrombosis risk in the general African American population in 327 SCD patients genotyped by the Affymetrix Pan-African Axiom GeneChip array. The median age of this cohort was 32 years (IQR, 23-43 years), 268 (82%) were Hb SS/Sβ0 genotype, 196 (60%) were female, and 153 (47%) were on hydroxyurea. Two previously reported variants in the thrombomodulin gene (THBD) were associated with thrombosis risk (THBD rs2567617: MAF, 0.25; odds ratio [OR], 1.50; 95% confidence interval, 1.01-2.22; P = .049; and THBD rs1998081: MAF, 0.24; OR, 1.46; 95% confidence interval; 0.99-2.17; P = .059). The MAF for the factor V Leiden and prothrombin gene mutations was <0.01, so we were unable to impute and analyze these variants in this cohort. The factor V Leiden and the prothrombin gene mutations were tested as part of routine medical care in 56 and 42 SCD patients, respectively, and neither mutation was detected.
Discussion
Thrombosis has been increasingly recognized as a complication of SCD that leads to substantial morbidity and early mortality.7-9,26 In a longitudinal cohort of 1193 SCD patients with a median follow-up of 6.4 years, we demonstrate that thrombotic events are common, with a cumulative incidence of 17.4%. We observed high rates of recurrence for both CVA (18%) and venous thrombotic events (33%) and that the use of direct factor Xa inhibitors was associated with a lower rate of VTE recurrence compared with other anticoagulation therapies.
Patients with SCD have a three- to 30-fold greater risk for CVA compared with the general African American population.27 A CVA occurred in 4% of our SCD patients, which is consistent with what has been observed in other cohorts.28,29 A lower Hb F% was associated with CVA risk in Hb SS/Sβ0-thalassemia patients, which has been observed in some SCD cohorts.4,30-32 We observed CVA recurrence in 18% of SCD patients, including a rate of 24% for CVA recurrence in those SCD patients that maintained at Hb fractionation goal for at least 80% of their laboratory checks. These rates are similar to the 18% recurrence rate described in SCD children at goal5 and highlights the need for alternative strategies to identify high-risk patients and for developing therapies to treat CVA in SCD.
SCD patients have a four- to 100-fold increased risk for VTE compared with the general population.33-35 Many of these cohorts have relied solely on ICD coding and the risk factors for VTE in SCD patients are not clear. We applied a comprehensive approach integrating VTE events, identified by ICD coding and manually confirmed through the medical records, with clinical and laboratory variables to identify risk factors for VTE. A VTE event occurred in 14% of our cohort, which is consistent with the 11% to 25% incidence reported in the literature.7-9
Decreasing eGFR was an independent risk factor for VTE in our cohort. In the general population, the risk for VTE is 1.3-fold higher in those with stage 1 CKD, increasing up to 2.1-fold higher in those with stage 3 or 4 CKD and 2.3-fold higher in those with stage 5 CKD.36 Several pathways implicated in CKD-related thrombosis, including increased activation of procoagulant factors and platelets, endothelial injury, and reduced anticoagulant and fibrinolytic activity, overlap with the vasculopathy of SCD and may highlight common pathways for CKD and thrombosis in SCD patients. A high white blood cell count and Hb concentration were also risk factors for VTE in our cohort. Both a higher white blood cell count and a higher Hb concentration are also risk factors for thrombosis in polycythemia vera.37,38 Common pathophysiologies, such as activated white blood cells promoting endothelial damage and a higher Hb concentration leading to hyperviscosity, may be involved in thrombosis risk for both polycythemia vera and SCD. Patients with SCD who developed a VTE in our cohort had more frequent hospitalizations, orthopedic surgeries, and central venous catheter placements. These risk factors, also identified in the general population39 and other SCD cohorts,6,8,9 may promote thrombosis by reflecting the severity of SCD, increasing venous stasis, and/or causing direct vascular injury.
In our study, hydroxyurea was associated with a greater risk for VTE, but not CVA, in the entire cohort as well as in the subset of Hb SS/Sβ0 patients and for non–catheter-related VTE. The use of hydroxyurea may be a marker for more severe SCD. Interestingly, hydroxyurea may have differential effects on the venous and arterial vasculature, as demonstrated in myeloproliferative neoplasms.40-42 In a study of patients with polycythemia vera, hydroxyurea conferred no protective benefit against VTE but led to a threefold reduction in arterial thrombotic events.40 Hydroxyurea targets multiple processes implicated in arterial thrombosis, such as qualitative changes in white blood cells, reduced expression of endothelial adhesion molecules, and enhanced nitric oxide generation.43 Reduced blood flow due to hyperviscosity is predominantly associated with venous thrombosis and may not be ameliorated by hydroxyurea.40 The association of hydroxyurea with VTE risk in SCD patients will need to be investigated in future studies.
VTE recurrence was observed in 33% of SCD patients and was lower in those treated with an oral direct factor Xa inhibitor (10%) compared with alternative therapies (warfarin, 39%; low-molecular-weight heparin, 38%). The advantage of direct oral anticoagulants over warfarin for thrombotic risk has been observed in some,44,45 but not all, non-SCD cohorts.46 This advantage may be partially dependent on the time in therapeutic range for warfarin.44 Interestingly, the proportion of INR results at goal was not associated with the rate of VTE recurrence. Future prospective studies comparing the safety and efficacy of direct oral anticoagulants vs warfarin to treat VTE in SCD are warranted.
Genetic variation may also contribute to the risk of thrombosis. Variants in ABO, IGFBP3, and THBD have been implicated in the thrombotic risk of African Americans.10 We tested these variants and were able to identify a 1.5-fold increased risk for thrombosis in SCD patients with the THBD rs2567617 and rs1998081 variants. THBD encodes an endothelial cell glycoprotein, thrombomodulin, that plays a central role in anticoagulation by (1) reducing circulating thrombin levels, (2) increasing the activation of protein C by >2 orders of magnitude after complexing with thrombin, and (3) downregulating complement activation.47,48 Variants in THBD have been implicated in both arterial49-53 and venous54-56 thrombotic complications as well as in the atypical hemolytic uremic syndrome.57 Future investigation for the effects of THBD rs2567617 and rs1998081 on thrombomodulin function and thrombotic risk in SCD may provide insight into the mechanisms and risks of SCD vasculopathy.
Limitations of this study include that it was a single-center study and retrospective in nature. Fragmented care is common in SCD patients,58 and our study was limited in the ability to detect thrombotic events that may have occurred at outside institutions, leading to an ascertainment bias. The median age at first encounter in SCD patients that developed a thrombotic event was older compared with those that did not have a thrombotic event, potentially leading to a selection bias. Orthopedic procedures, central venous catheter procedures, and pregnancies were captured regardless of timing to thrombosis, limiting our interpretation to associations, not causation. The definition of hydroxyurea was ever vs never in this study. The use and adherence of hydroxyurea among eligible patients is low,59 and future prospective studies capturing the timing of procedures and pregnancies and confirming hydroxyurea use will help address these limitations.
In summary, in a cohort of nearly 1200 SCD patients, we observed a high rate of thrombotic events and identified several traditional (hospitalization rate, central venous catheter placement, pregnancy, and kidney disease) and SCD-specific (hydroxyurea therapy, Hb SS/Sβ0-thalassemia genotype, lower Hb F%, higher AST, higher white blood cell count, and higher Hb concentration) risk factors for thrombosis. In addition, we demonstrated a very high rate of recurrence for CVA and venous thrombotic events and that SCD patients treated with a direct oral factor Xa inhibitors had lower rates of VTE recurrence compared with other treatment strategies. Future prospective studies understanding the role of thrombomodulin in maintaining vascular flow and developing thrombotic risk profiles may guide the prophylactic and treatment strategies for SCD.
For data sharing, e-mail the corresponding author, Santosh L. Saraf (e-mail: ssaraf@uic.edu).
Acknowledgments
The project described was supported by the National Institutes of Health, National Heart, Lung, and Blood Institute through grants K23 HL125984 and R03 HL146788 (S.L.S).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: A.S., R.R., X.Z., and S.L.S. designed and performed research, analyzed the data, and wrote the paper; and B.N.S., J.H., M.G., R.E.M., and V.R.G. designed and performed research and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Santosh L. Saraf, Division of Hematology & Oncology, Department of Medicine, University of Illinois at Chicago, 820 S Wood St, MC 712, Chicago, IL 60612; e-mail: ssaraf@uic.edu.

