

Abstract
Acute myeloid leukemia (AML) is a highly heterogeneous disease arising from acquired genetic and epigenetic aberrations which stifle normal development and differentiation of hematopoietic precursors. Despite the complex and varied biological underpinnings, induction therapy for AML has remained fairly uniform over 4 decades and outcomes remain poor for most patients. Recently, enhanced understanding of the leukemic epigenome has resulted in the translational investigation of a number of epigenetic modifying agents currently in various stages of clinical development. These novel therapies are based on mechanistic rationale and offer the potential to improve AML patient outcomes. In light of many recent advances in this field, we provide an updated, clinically oriented review of the evolving landscape of epigenetic modifying agents for the treatment of AML.
Introduction
Epigenetic alterations constitute a series of heritable, yet modifiable, molecular changes that modulate gene expression without discrete mutations in the genes themselves. In acute myeloid leukemia (AML), recurring genetic mutations, such as FLT3, CEBPA, and NPM1, fail to fully account for the extensive molecular and clinical heterogeneity. Indeed, myeloid precursors accumulate genetic mutations and epigenetic alterations that impair normal maturation and impart the ability to evade apoptosis and replicate indefinitely. Some epigenetic abnormalities result directly or indirectly from mutations in epigenetic regulators, such as DNMT3A, IDH, and TET2. However, in recent years, the use of epigenetic profiling has also defined recurring methylation patterns representing prognostically significant AML subtypes distinct from previously recognized genetic subtypes.1,2 Furthermore, the epigenetic complexity of AML increases as the disease progresses, and elderly AML patients accumulate these defects at a greater frequency among genes associated with myeloid differentiation.3,4
Traditionally, myeloablative cytotoxic chemotherapy, often followed by hematopoietic stem cell transplantation, has been the standard of care for AML, but common treatment-related toxicities exclude many patients. Poor outcomes in elderly and unfit patients have necessitated alternative treatment strategies. With the recognition of this unmet clinical need, in conjunction with an appreciation of the fundamental epigenetic underpinnings of AML, a therapeutic class, collectively referred to as “epigenetic modifying agents,” has emerged as a mechanism-based low-intensity therapeutic approach for AML patients incorporated as front-line and salvage therapy, although few agents have received US Food and Drug Administration approval for AML.
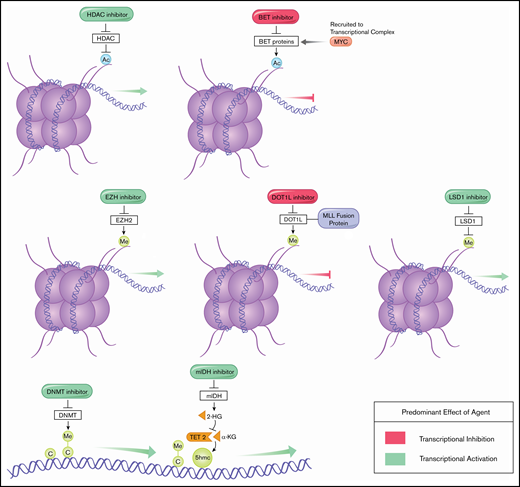
Epigenetic modifying agents serve as a moderately effective but often more tolerable alternative to intensive chemotherapy which has historically produced unacceptably high mortality rates in the elderly.5 Azacitidine, for example, subjects patients to less bone marrow suppression, nausea, and febrile neutropenia than intensive chemotherapy and is well-tolerated by elderly patients.6 Additionally, the ability of these agents to modify gene expression (Figure 1) and reverse malignant adaptations offers the potential for synergistic pairings. Over the last 2 decades, certain epigenetic therapies have entered standard care, whereas others in various stages of development show therapeutic potential. Here, we review the successes and limitations of the expanding landscape of epigenetic modifiers in the treatment of AML and expectations from this class moving forward.
DNA methyltransferase inhibitors
DNA methyltransferase (DNMT) methylates cytosine residues within cytosine guanine dinucleotide islands, preventing transcription factors from binding promoter regions and, thereby, silencing gene expression. Aberrant DNA methylation can silence genes involved in differentiation, DNA repair, and apoptosis and is a major driver for the development of myelodysplastic syndrome (MDS) and the progression of MDS to AML.7
Hypomethylating agents (HMAs) reverse dysregulated DNA methylation and are the most well-studied epigenetic therapies. The 2 most notable HMAs, 5-azacytidine (azacitidine) and 5-aza-2′-deoxycytidine (decitabine), are cytosine analogs. When activated, decitabine becomes incorporated into the DNA of myeloblast progeny and binds irreversibly to DNMTs, which are then degraded through proteasomal pathways.8 About 10% to 20% of azacitidine is also incorporated into DNA, whereas the majority becomes incorporated into RNA and disrupts messenger RNA and protein synthesis.9 Thus, although both agents exert DNMT-depleting effects, decitabine does so at levels twofold to 10-fold lower than azacitidine.10,11 The resulting DNA hypomethylation promotes reactivation of silenced tumor suppressor genes in vitro and is theorized to induce cellular differentiation and apoptosis.12-14 However, confirming these postulated mechanisms in patients has been difficult, and clinical studies have focused on demethylation of the tumor suppressor p15 and global demethylation in response to HMA treatment.15,16 Because HMAs are known to have a second mechanism of action with direct cytotoxicity at higher doses, the relative contributions of DNA demethylation and cellular cytotoxicity have not been clearly parsed out.10,17 Further studies are needed to better understand the clinically relevant mechanisms of action of HMAs.16
Azacitidine and decitabine have found success among elderly AML patients and certain high-risk subsets, including patients with chromosome 5 and 7 abnormalities who often fare better with HMA therapy than with conventional care.18,19 Both agents were initially evaluated in MDS, and early data showing their efficacy in AML came from a 2009 post hoc analysis of the AZA-MDS-001 trial, in which a subset of intermediate- and high-risk MDS cases were classified as low blast count AML (20%-30% blasts). Among the 113 elderly patients identified with low blast count AML, randomization to azacitidine significantly improved overall survival (OS) over conventional care, with medians of 24.5 months and 16.0 months, respectively (P = .005).19 These results paved the way for trials designed to investigate HMAs as frontline therapy in AML.
In a phase 3 study in 2012, 485 patients older than 65 years with newly diagnosed AML ineligible for intensive chemotherapy were randomized to decitabine or conventional treatment, consisting of supportive care or low-dose cytarabine.20 The study found a significant increase in complete response (CR) rate with decitabine compared with conventional treatment (17.8% vs 7.8%; P = .001). Furthermore, decitabine was tolerable; the most common grade 3/4 adverse events (AEs) were thrombocytopenia (27%) and neutropenia (24%).20 Unfortunately, failure to demonstrate significantly increased OS at the time of planned analysis prevented the approval of decitabine for AML in the United States. That same year, Quintás-Cardama et al published a retrospective study of a more fit population of 671 patients older than 65 years treated with intensive chemotherapy or azacitidine/decitabine-based therapy.21 The investigators noted higher CR rates with chemotherapy than with HMA-based therapy but similar 2-year relapse-free survival rates and median OS. Together, these studies demonstrated that azacitidine and decitabine were safe and effective treatment options for elderly patients with newly diagnosed AML (notable monotherapy trials summarized in Table 1).
Major monotherapy trials of epigenetic-directed therapies
| Class | Agent | Trial phase | Target population | Patients, n | Response rate | Identifier |
|---|---|---|---|---|---|---|
| HMA | Decitabine vs treatment choice | 3 | Newly diagnosed AML ineligible for intensive chemotherapy | 485 | 17.8% vs 78% CR/CRp, respectively. | NCT00260832 |
| HMA | Azacitidine/decitabine vs intensive chemotherapy | Retrospective* | Newly diagnosed AML | 671 | 28% vs 42% CR, respectively† | NCT00926731, NCT00952588 |
| HMA | Guadecitabine | 2 | R/R AML | 103 | 23% CR/CRi | NCT01261312 |
| HMA | Guadecitabine | 2 | Treatment-naive AML ineligible for intensive chemotherapy | 107 | 57% CR/CRi/CRp | NCT01261312 |
| HMA | Azacitidine vs conventional care | 3 | Newly diagnosed AML with 20-30% blasts ineligible for intensive chemotherapy | 113 | 18% vs 16% CR, respectively (P = .80). | NCT00071799 |
| HMA | Azacitidine vs conventional care | 3 | Newly diagnosed AML with > 30% blasts ineligible for intensive chemotherapy | 488 | 27.8% vs 25.1% CR/CRi, respectively (P = 0.54)‡ | NCT01074047 |
| HDACi | Vorinostat | 2 | R/R AML or newly diagnosed AML ineligible for chemotherapy | 37 | 2.7% CR | NCT00305773 |
| mIDH inhibitor | Enasidenib | 1/2 | IDH2-mutated R/R AML | 239 | 19.3% CR, 6.8% CRi/CRp | NCT01915498 |
| mIDH inhibitor | Ivosidenib | 1 | IDH1-mutated R/R AML | 125 | 21.6% CR, 8.8% CRh | NCT02074839 |
| BET inhibitor | GSK525762 | 1 | R/R AML | 46 | 2.2% CRi, 2.2% CRp | NCT01943851 |
| BET inhibitor | ABBV-075 (mivebresib) | 1 | R/R AML | 19 | 5.3% CRp, preliminary data | NCT02391480 |
| BET inhibitor | OTX015 | 1 | R/R AML and AML ineligible for intensive chemotherapy | 36 | 2.8% CR, 33.3% CRp | NCT01713582 |
| DOT1L inhibitor | EPZ-5676 (pinometostat) | 1 | Pediatric R/R MLL rearranged AML | 18 | No complete responses observed | NCT02141828 |
| Class | Agent | Trial phase | Target population | Patients, n | Response rate | Identifier |
|---|---|---|---|---|---|---|
| HMA | Decitabine vs treatment choice | 3 | Newly diagnosed AML ineligible for intensive chemotherapy | 485 | 17.8% vs 78% CR/CRp, respectively. | NCT00260832 |
| HMA | Azacitidine/decitabine vs intensive chemotherapy | Retrospective* | Newly diagnosed AML | 671 | 28% vs 42% CR, respectively† | NCT00926731, NCT00952588 |
| HMA | Guadecitabine | 2 | R/R AML | 103 | 23% CR/CRi | NCT01261312 |
| HMA | Guadecitabine | 2 | Treatment-naive AML ineligible for intensive chemotherapy | 107 | 57% CR/CRi/CRp | NCT01261312 |
| HMA | Azacitidine vs conventional care | 3 | Newly diagnosed AML with 20-30% blasts ineligible for intensive chemotherapy | 113 | 18% vs 16% CR, respectively (P = .80). | NCT00071799 |
| HMA | Azacitidine vs conventional care | 3 | Newly diagnosed AML with > 30% blasts ineligible for intensive chemotherapy | 488 | 27.8% vs 25.1% CR/CRi, respectively (P = 0.54)‡ | NCT01074047 |
| HDACi | Vorinostat | 2 | R/R AML or newly diagnosed AML ineligible for chemotherapy | 37 | 2.7% CR | NCT00305773 |
| mIDH inhibitor | Enasidenib | 1/2 | IDH2-mutated R/R AML | 239 | 19.3% CR, 6.8% CRi/CRp | NCT01915498 |
| mIDH inhibitor | Ivosidenib | 1 | IDH1-mutated R/R AML | 125 | 21.6% CR, 8.8% CRh | NCT02074839 |
| BET inhibitor | GSK525762 | 1 | R/R AML | 46 | 2.2% CRi, 2.2% CRp | NCT01943851 |
| BET inhibitor | ABBV-075 (mivebresib) | 1 | R/R AML | 19 | 5.3% CRp, preliminary data | NCT02391480 |
| BET inhibitor | OTX015 | 1 | R/R AML and AML ineligible for intensive chemotherapy | 36 | 2.8% CR, 33.3% CRp | NCT01713582 |
| DOT1L inhibitor | EPZ-5676 (pinometostat) | 1 | Pediatric R/R MLL rearranged AML | 18 | No complete responses observed | NCT02141828 |
2-HG, 2-hydroxyglutarate; 5hmc, 5-hydroxymethylcytosine; α-KG, α-ketoglutarate; Ac, acetyl; BET, bromodomain extraterminal protein; C, cytosine; CRh, CR with partial hematologic recovery; CRp, CR with incomplete platelet recovery; DNMT, DNA methyltransferase; DOT1L, disruptor of telomeric silencing 1-like; EZH, enhancer of zeste homolog; HDAC, histone deacetylase; LSD1, lysine-specific demethylase 1; Me, methyl; mIDH, mutant isocitrate dehydrogenase 1; MLL, mixed lineage leukemia.
One retrospective study was included that had significant implications for AML treatment.
Despite lower CR, median survival was similar compared with intensive chemotherapy.
Despite similar CR, median survival was higher with azacitidine.
Azacitidine has also shown potential as maintenance postremission therapy. In a phase 3 study with 116 AML/MDS patients who attained CR or CR with insufficient count recovery (CRi) after chemotherapy, azacitidine maintenance improved disease-free survival compared with placebo (15.9 vs 10.3 months; P = .04).22 Subsequent results from the larger QUAZAR study of 472 AML patients in first CR/CRi also found a significant improvement in OS with oral azacitidine (CC-486) maintenance vs placebo (24.7 vs 14.8 months, respectively; P = .0009).23,24
Guadecitabine is a next-generation HMA that was rationally designed to resist degradation by cytidine deaminase by linking decitabine, its active metabolite, to deoxyguanosine by a phosphodiester bond. This modification prolongs its exposure window and may improve marrow penetration. Early studies have demonstrated greater levels of DNA hypomethylation in vivo with guadecitabine therapy than were previously seen with azacitidine or decitabine.25 In a phase 2 study evaluating guadecitabine in 107 treatment-naive or relapsed or refractory (R/R) AML patients, the most common AEs were febrile neutropenia (61%), thrombocytopenia (49%), and anemia (29%).26,27 Disappointingly, in a report on ASTRAL-1, a subsequent randomized-controlled trial (RCT) of guadecitabine vs physician’s choice of azacitidine, decitabine, or low-dose cytarabine in treatment-naive AML, guadecitabine failed to improve upon CR (19.4% vs 17.4%, respectively; P = .48) or OS (7.1 vs 8.47 months; P = .73).28 A phase 3 study comparing guadecitabine with treatment choice in R/R AML is ongoing (NCT02920008), as is a study of guadecitabine with idarubicin (NCT02096055).
Currently, initiation of treatment with azacitidine or decitabine requires a commitment to clinic visits 5 to 7 days per month for subcutaneous or IV administration, respectively. Oral HMAs have been developed to improve patient convenience and facilitate longer-term administration with the hopes of optimizing drug exposure. An oral formulation of azacitidine, CC-486, has demonstrated clinical activity and safety in a phase 1 study of patients with AML, MDS, or chronic myelogenous leukemia (CML), as well as in a phase 2 study of patients with lower-risk MDS.29,30 A phase 3 study comparing oral azacitidine plus supportive care with supportive care alone in transfusion-dependent lower-risk MDS is ongoing (NCT01566695). A second oral agent targets the rapid metabolism of HMAs in the gut and liver by cytidine deaminase, which ordinarily limits their oral bioavailability. ASTX727, a novel combination of the selective cytidine deaminase inhibitor cedazuridine and oral decitabine, achieved equivalent area under the curve exposure to decitabine and had comparable efficacy and safety in phase 2 and 3 studies of patients with myeloid malignancies.31,32
Despite the proven utility of HMA monotherapy as a low-intensity treatment option in AML, there remains a need to attain higher response rates and durability of response, because relapse of disease eventually occurs in essentially all cases with poor outcomes after HMA failure.33 Furthermore, HMAs require 3 or 4 cycles to achieve best response, and interruption in treatment is associated with rapid loss of response.34 Increasingly, HMAs have been evaluated in combination with other agents to address these shortcomings. The selective BCL2 inhibitor venetoclax has limited single-agent activity in AML, with a major determinant of venetoclax resistance being adaptive upregulation of the closely related antiapoptotic protein MCL-1.35 In preclinical studies, azacitidine downregulated MCL-1 and synergistically induced apoptosis when combined with venetoclax.36 The combination of HMAs and venetoclax demonstrated safety and efficacy in a phase 1 study and has been granted accelerated approval by the US Food and Drug Administration for treatment-naive AML patients ineligible for intensive chemotherapy.37 With 145 elderly treatment-naive patients treated, 67% achieved CR or CRi, which compares favorably with historical response rates with HMA monotherapy.6,20 Notably, median time to best response was 1.8 months compared with 4.3 months with decitabine and 3.5 months with azacitidine.20,38 A phase 2 trial of the combination is ongoing (NCT03466294). HMAs also increase the expression of tumor-related genes that can sensitize malignant cells to immune recognition and attack. In AML patients treated with azacitidine, upregulation of the program death pathway (programmed cell death protein-1 [PD-1]/programmed death ligand-1) blocks cytotoxic T-cell activity and has been associated with azacitidine resistance.39 Considering these findings, Daver et al26 demonstrated the safety of azacitidine and nivolumab in AML in a phase 1 study, with immune-mediated toxicities (ie, pneumonitis, nephritis) occurring in 25% of patients. A phase 2 trial of this combination in R/R AML and treatment-naive AML is underway; preliminary data showed CR/CRi in 18% of treated patients with relapsed AML (NCT02397720).26
The identification of effective biomarkers of HMA response has been another crucial area of study. Potential genetic and molecular predictors of favorable responses to HMAs include the presence of p53, IDH, TET2, and DNMT3A mutations, normal lactate dehydrogenase (LDH), and elevated fetal hemoglobin levels.40-45 Clinically, male sex, older age, and lower performance score portend poor responses to HMAs.46-48 In MDS, bone marrow blasts >15%, abnormal karyotype, and previous cytarabine treatment predicted poor HMA response, whereas factors including performance status ≥2, circulating blasts, and red blood cell transfusion dependency ≥4 units in 8 weeks portend poor OS.49
Histone deacetylase inhibitors
Acetylation of lysine residues by histone acetyltransferases promotes relaxation of chromatin and exposure of DNA to transcription factor binding. Conversely, removal of acetyl groups by histone deacetylase (HDAC) renders DNA less accessible to transcription factors. The disruption of this balance in favor of histone hypoacetylation plays a crucial role in leukemogenesis by contributing to repression of genes critical for normal cellular development.50
HDAC inhibitors (HDACi’s) were developed to interfere with this process and restore normal histone acetylation patterns, inducing expression of genes that promote proliferation arrest, differentiation, and apoptosis in cancer cells.51 HDACi’s also increase the acetylation of nonhistone proteins, such as heat shock protein 90, a chaperone that protects oncogenic client proteins from proteasome-directed degradation. Heat shock protein 90 acetylation interferes with its chaperone function, disrupting oncogenic signaling pathways.52 At pharmacologic doses, HDACi’s also directly induce double-strand breaks and oxidative DNA damage in leukemic cells.53
Vorinostat is an HDACi that rapidly induces histone acetylation in leukemic blasts in the peripheral blood and the bone marrow but has produced disappointing clinical results. Anthracyclines induce double-stranded DNA breaks adjacent to histones and, thus, were predicted to have synergy with HDACi’s.54 However, a phase 3 study did not show any difference in outcomes when vorinostat was added to cytarabine and idarubicin.55 In vitro studies identified vorinostat and azacitidine as another promising combination, with a synergistic effect on reexpression of downregulated genes in cancer cells lines, including those encoding immunogenic antigens presented on MHC class I molecules.56,57 Despite encouraging early clinical data for the combination, a subsequent 2017 RCT comparing vorinostat plus azacitidine with azacitidine alone in AML and high-risk MDS found no difference in overall response rate (42% and 41%, respectively).58 The same year, North American Intergroup Study S1117 was published; it randomized higher-risk MDS and chronic myelomonocytic leukemia (CMML) patients 1:1:1 to azacitidine alone, azacitidine plus vorinostat, or azacitidine plus lenalidomide. Similarly, no benefit to combination therapy was found in MDS, although the addition of lenalidomide improved survival in CMML.59
Panobinostat, an oral pan-deacetylase inhibitor that is >10 times more potent than vorinostat, produced significant results in multiple myeloma and showed preclinical synergy with HMAs in AML.60,61 As with vorinostat, early data using panobinostat and azacitidine showed tolerability and clinical activity of the combination.62 However, a phase 2b trial randomizing 82 patients with MDS, CML, or AML to panobinostat and azacitidine or azacitidine alone found that patients derived no benefit from the addition of panobinostat.63 No differences were observed in response rates or OS; in fact, patients receiving the combination experienced more grade ≥3 AEs (97.4% vs 81%) and on-treatment deaths (13.2% vs 4.8%) than did those receiving azacitidine alone.
The unfulfilled preclinical promise of the HMA-HDACi combination may relate, in part, to the potential of HDACi’s for pharmacodynamic antagonism of HMAs.64 Different scheduling regimens will need to be explored in the future to attempt to circumvent this issue. It was also hypothesized that the relative impotency of vorinostat compared with other HDACi’s may have limited its synergistic potential, although the negative studies with panobinostat have weakened this argument. Several other HDACi’s are currently being investigated, including novel agents like pracinostat, entinostat, and romidepsin. Early trials of these agents, in combination with azacitidine, have shown clinical activity and safety (Table 2).65,66 However, as experience has demonstrated, RCTs are needed to discern their true benefit in AML.
Major monotherapy trials of epigenetic-directed therapies
| Class | Agent | Trial phase | Target population | Patients, n | Response | Identifier |
|---|---|---|---|---|---|---|
| HMA + BCL2 inhibitor | Azacitidine + venetoclax | 1 | Elderly, treatment-naive AML | 145 | 67% CR/CRi | NCT02203773 |
| HMA + anti–PD-1 | Azacitidine + nivolumab | 1b/2 | R/R AML | 51 | 18% CR/CRi | NCT02397720 |
| HMA + chemotherapy | Azacitidine + chemotherapy vs chemotherapy alone | 3 | Newly diagnosed AML | 209 | 48% and 52% CR, respectively | NCT00915252 |
| HMA + mIDH inhibitor | Azacitidine + enasidenib or ivosidenib | 1b/2* | IDH-mutated newly diagnosed AML ineligible for intensive chemotherapy | 13 | 33% CR enasidenib, 43% CR ivosidenib | NCT02677922 |
| HDACi + chemotherapy | Vorinostat + idarubicin and cytarabine | 2 | Newly diagnosed AML or higher-risk MDS | 75 | 76% CR and 9% CRp | NCT00656617 |
| HMA + HDACi | Vorinostat + azacitidine | 1 | AML or MDS | 6 AML, 14 MDS | 45% CR and 9% CRi | NCT00392353 |
| HMA + HDACi | Vorinostat + azacitidine vs azacitidine alone | 2 | AML and high-risk MDS ineligible for intensive chemotherapy | 217 AML, 42 MDS | 26% and 22% CR/CRi/mCR, respectively (P = .49)† | NCT01617226 |
| HMA + HDACi | Azacitidine + pracinostat | 2 | Newly diagnosed AML ineligible for intensive chemotherapy | 50 | 42.0% CR | NCT01912274 |
| HMA + HDACi | Romidepsin + azacitidine | 1 | Newly diagnosed or R/R AML ineligible for intensive chemotherapy | 46 | 38.9% CR/CRi | ISRCTN69211255 |
| mIDH inhibitor + chemotherapy | Enasidenib or ivosidenib + chemotherapy | 1 | Newly diagnosed IDH1- or IDH2-mutated AML | 65 | 69.6% CR/CRi/CRp (ivosidenib) and 62.2% CR/CRi/CRp (enasidenib) | NC02632708 |
| Class | Agent | Trial phase | Target population | Patients, n | Response | Identifier |
|---|---|---|---|---|---|---|
| HMA + BCL2 inhibitor | Azacitidine + venetoclax | 1 | Elderly, treatment-naive AML | 145 | 67% CR/CRi | NCT02203773 |
| HMA + anti–PD-1 | Azacitidine + nivolumab | 1b/2 | R/R AML | 51 | 18% CR/CRi | NCT02397720 |
| HMA + chemotherapy | Azacitidine + chemotherapy vs chemotherapy alone | 3 | Newly diagnosed AML | 209 | 48% and 52% CR, respectively | NCT00915252 |
| HMA + mIDH inhibitor | Azacitidine + enasidenib or ivosidenib | 1b/2* | IDH-mutated newly diagnosed AML ineligible for intensive chemotherapy | 13 | 33% CR enasidenib, 43% CR ivosidenib | NCT02677922 |
| HDACi + chemotherapy | Vorinostat + idarubicin and cytarabine | 2 | Newly diagnosed AML or higher-risk MDS | 75 | 76% CR and 9% CRp | NCT00656617 |
| HMA + HDACi | Vorinostat + azacitidine | 1 | AML or MDS | 6 AML, 14 MDS | 45% CR and 9% CRi | NCT00392353 |
| HMA + HDACi | Vorinostat + azacitidine vs azacitidine alone | 2 | AML and high-risk MDS ineligible for intensive chemotherapy | 217 AML, 42 MDS | 26% and 22% CR/CRi/mCR, respectively (P = .49)† | NCT01617226 |
| HMA + HDACi | Azacitidine + pracinostat | 2 | Newly diagnosed AML ineligible for intensive chemotherapy | 50 | 42.0% CR | NCT01912274 |
| HMA + HDACi | Romidepsin + azacitidine | 1 | Newly diagnosed or R/R AML ineligible for intensive chemotherapy | 46 | 38.9% CR/CRi | ISRCTN69211255 |
| mIDH inhibitor + chemotherapy | Enasidenib or ivosidenib + chemotherapy | 1 | Newly diagnosed IDH1- or IDH2-mutated AML | 65 | 69.6% CR/CRi/CRp (ivosidenib) and 62.2% CR/CRi/CRp (enasidenib) | NC02632708 |
IDH, isocitrate dehydrogenase; mCR, marrow CR.
Cited data are from preliminary results.
No difference in survival was noted.
Isocitrate dehydrogenase inhibitors
Isocitrate dehydrogenase 1 (IDH1) and IDH2, enzymes essential to the maintenance of normal energy balance, catalyze the metabolism of isocitrate to α-ketoglutarate (α-KG), an important cofactor for histone demethylase and 5-methylcytosine hydroxylase. IDH mutations are found in several malignancies, including AML and gliomas, and confer a gain-of-function whereby mutant IDH converts α-KG to the oncometabolite 2-hydroxyglutarate (2-HG). The chemical similarity of 2-HG to α-KG allows it to competitively inhibit α-KG–dependent enzymes, including histone demethylase and TET2, an enzyme that converts 5-methylcytosine to 5-hydroxymethylcytosine. The decreased activity of enzymes like TET2 results in a widespread increase in histone and DNA methylation, as well as changes in 5-hydroxymethylcytosine levels that block normal myeloid differentiation.67,68 Additionally, loss of TET2 function synergizes with FLT3ITD mutations to promote further hypermethylation and leukemogenesis.69
Overall, IDH mutations are seen in ∼16% of AML cases but are particularly common in secondary AML, having been linked to the transformation of myeloproliferative neoplasms (MPNs) and MDS to AML.70 Data regarding the prognostic significance of IDH mutations in AML have been conflicting and may depend on the specific point mutation involved, as well as the presence or absence of comutations.71
Ivosidenib and enasidenib are selective inhibitors of mutant IDH1 and IDH2, respectively. Stein et al evaluated the use of enasidenib in IDH2-mutated R/R AML in a phase 1/2 study. One quarter of the 109 treated patients attained CR/CRi.72 Grade 3/4 AEs included hyperbilirubinemia (12%) and differentiation syndrome (7%). After the first treatment cycle, plasma 2-HG levels were reduced 93% in patients with IDH2-R140Q mutations and 28% in patients with IDH2-R172K mutations. Bone marrow aspirates from responders showed a reduction in myeloblast percentage and an emergence of mature myeloid forms retaining IDH2 mutations, indicating differentiation from malignant blasts. Nonresponders harbored higher overall comutational burden and with a specific predilection for RAS pathway mutations.73 Based on these results, enasidenib was approved as monotherapy for R/R IDH2-mutated AML.
Ivosidenib was first approved for R/R IDH1-mutated AML and, more recently, for newly diagnosed IDH1-mutated AML in patients older than 75 years of age or ineligible for intensive chemotherapy. Initial approval was based on a phase 1 trial treating patients with advanced hematologic malignancies with ivosidenib.74 Of 125 R/R AML patients, CR was attained in 21.6% for a median of 9.3 months, whereas CR with incomplete hematologic recovery was attained in 8.8%. Grade 3/4 AEs included QT prolongation (7.8%), thrombocytopenia (3.4%), and differentiation syndrome (3.4%). As with enasidenib, greater comutational burden was associated with poorer response, although no specific genes, including NRAS, were of particular significance. Approval was expanded to first-line therapy for IDH1-mutated AML patients older than 75 years of age or otherwise unfit for intensive chemotherapy after ivosidenib resulted in CR in 30% of elderly patients newly diagnosed with AML.75
Both of these agents are now being investigated in combination with chemotherapy and HMAs as part of upfront treatment of IDH-mutated AML. A recent phase 1 trial investigated the combination of intensive chemotherapy and ivosidenib or enasidenib in 134 patients with newly diagnosed IDH1- or IDH2-mutated AML.76 In de novo AML, the combination achieved a CR, CRi, or CR with incomplete platelet recovery (CRp) rate in 93% of patients taking ivosidenib and 73% of patients taking enasidenib, whereas rates in secondary AML were 46% and 63%, respectively. The combination was tolerable, and the most frequent grade 3/4 AE was febrile neutropenia, which was seen in just over half of treated patients. A phase 3 trial is planned to determine the benefit of the addition of ivosidenib and enasidenib.
A preclinical study combining ivosidenib and azacitidine in a mutant-IDH1 cell model found a synergistic effect on the induction of differentiation and cell death.77 An ongoing phase 1b/2 study demonstrated the tolerability and efficacy of azacitidine with ivosidenib or enasidenib in 11 patients with newly diagnosed IDH-mutated AML, with rates of hematologic AEs similar to historical rates with azacitidine alone.78 Encouragingly, responses, including 4 CRs, were observed in 8 of 11 patients in this small sample. Preliminary results from the randomized phase 2 study of enasidenib and azacitidine have shown superior rates and depths of response with the combination compared with azacitidine alone in newly diagnosed AML (overall response rate 68% vs 42%, respectively; P = .0155; and CR, 50% vs 12%; P = .0002).79 Enrollment is ongoing in a phase 3 study of ivosidenib and azacitidine (NCT03173248).
Bromodomain inhibitors
Bromodomain inhibitors constitute a more recent class of epigenetic modifiers being investigated in AML. The diverse family of bromodomain and extraterminal (BET) proteins possesses 2 N-terminal bromodomains, highly conserved 110-aa domains that bind to acetylated lysine residues on histones and other nuclear proteins to initiate transcriptional complexes.80 Although histone acetylation itself promotes transcriptional activation, BET proteins add an additional layer of transcriptional regulation.81
Bromodomain-containing 4 (BRD4) is 1 member of the BET family of proteins of particular interest in cancer that is a result, in part, of its role in mitotic progression.81 BRD4-initiated transcriptional complexes increase the expression of several proto-oncogenes, including MYC, BCL2, and CDK6, which promote self-renewal and maintain malignant cells in an undifferentiated state.82,83 In 2011, Zuber et al found that small hairpin RNA knockdown of BRD4 expression in mouse AML models had potent antileukemic effects, solidifying the protein as an enticing target in AML.84
OTX015 (MK-8628) is a novel oral agent that binds specifically to BET proteins BRD2, BRD3, and BRD4. By blocking their ability to bind to acetylated histones and activate transcription, OTX015 induces cell cycle arrest and apoptosis in AML cell lines.85 A phase 1 dose-escalation study evaluated doses from 10 to 160 mg daily in 36 patients with R/R AML, 16 cases of which were secondary AML.86 At 120 mg daily, grade 1-2 diarrhea and rashes hampered compliance. In total, 2 AML patients attained CR/CR with incomplete platelet recovery and 2 others had partial blast clearance.86 Investigators chose a dose of 80 mg daily for phase 2.
Several other BET inhibitors (BETi’s) have been developed recently and are being investigated in phase 1/2 studies (Table 3). Initial phase 1 data have generally shown modest efficacy with BETi monotherapy.86-89 However, preclinical findings demonstrate enhanced anticlonal in vitro activity as combination therapy, with HDACi’s, HMAs, and BCL2 and MCL1 inhibitors compared to monotherapy.85,90-92
Recent or ongoing trials of epigenetic-directed therapies
| Class | Agent | Phase | Target population | Status | Identifier |
|---|---|---|---|---|---|
| HMA | Guadecitabine | 3 | R/R AML | Active, not recruiting | NCT02920008 |
| HMA + DLI | Guadecitabine + DLI | 2 | AML or MDS relapsed post-AlloSCT | Recruiting | NCT02684162 |
| HMA | Guadecitabine | 3 | Newly diagnosed AML ineligible for intensive chemotherapy | Completed | NCT02348489 |
| HMA ± chemotherapy | Guadecitabine ± idarubicin | 2 | AML age ≥70 y | Active, not recruiting | NCT02096055 |
| HMA | Guadecitabine | 2 | AML with 20%-30% blasts or MDS refractory to azacitidine | Completed | NCT02197676 |
| HMA | Guadecitabine | 1/2 | AML, MDS | Completed | NCT01261312 |
| HMA + anti–PD-1 | Guadecitabine + atezolizumab | 1 | AML | Suspended | NCT02892318 |
| HDACi | Entinostat | 2 | AML age ≥60 y | Recruiting | NCT01305499 |
| HDACi + HMA | Entinostat + azacitidine | 1 | AML, MDS, CML | Active, not recruiting | NCT00101179 |
| HDACi + chemotherapy | Panobinostat + cytarabine and daunorubicin | 1 | AML, MDS age ≥60 | Active, not recruiting | NCT01463046 |
| HDACi + chemotherapy | Panobinostat + cytarabine and idarubicin | 1/2 | AML age ≥65 y | Completed | NCT00840346 |
| HDACi + chemotherapy | Panobinostat + cytarabine and idarubicin | 1 | AML age <65 y | Completed | NCT01242774 |
| HDACi + HMA | Panobinostat + azacitidine | 1 | AML <30% blasts, MDL, CMML, | Active, not recruiting | NCT00946647 |
| HDACi + HMA | Panobinostat + decitabine | 1/2 | AML age ≥60, MDS | Completed | NCT00691938 |
| HDACi + HMA | AR-42 + decitabine | 1 | AML | Completed | NCT01798901 |
| HDACi | Belinostat | 2 | AML age ≥60 | Completed | NCT00357032 |
| HDACi + HMA | Mocetinostat + azacitidine | 1/2 | AML, MDS | Completed | NCT00324220 |
| HDACi | Romidepsin | 2 | R/R AML | Completed | NCT00062075 |
| mIDH1 inhibitor | BAY1436032 | 1 | mIDH1 AML | Completed | NCT03127735 |
| mIDH1 inhibitor | Ivosidenib + venetoclax | 1/2 | mIDH1 AML, MDS/MPN | Recruiting | NCT03471260 |
| mIDH1 inhibitor ± HMA, chemotherapy | FT-2102 ± azacitidine or cytarabine | 1/2 | mIDH1 AML, MDS | Recruiting | NCT02719574 |
| mIDH2 inhibitor + HMA | Enasidenib + azacitidine | 2 | R/R mIDH2 AML | Recruiting | NCT03683433 |
| mIDH1, mIDH2 inhibitors + chemotherapy | Ivosidenib or enasidenib + cytarabine and daunorubicin or idarubicin | 1 | Newly diagnosed mIDH1 or mIDH2 AML | Active, not recruiting | NCT02632708 |
| mIDH1, mIDH2 inhibitors + HMA | Ivosidenib or enasidenib + azacitidine | 1/2 | Newly diagnosed mIDH1 or mIDH2 AML ineligible for intensive chemotherapy | Active, not recruiting | NCT02677922 |
| mIDH1 inhibitor + HMA | Ivosidenib or placebo + azacitidine | 3 | Newly diagnosed mIDH1 AML | Recruiting | NCT03173248 |
| BETi | OTX015 (MK-8628) | 1 | AML, ALL, NHL, MM | Completed | NCT01713582 |
| BETi ± HMA | FT-1101 ± azacitidine | 1 | R/R AML | Recruiting | NCT02543879 |
| BETi | GSK525762 | 1 | AML, NHL, MM | Recruiting | NCT01943851 |
| BETi | INCB054329 | 1/2 | AML, MDS/MPN, MM, solid tumor, or lymphoma | Completed | NCT02431260 |
| BETi ± BCL2 inhibitor | ABBV-075 +/− venetoclax | 1 | AML, MM, solid tumors, NHL | Active, not recruiting | NCT02391480 |
| EZH 1/2 inhibitor | DS-3201b | 1 | AML or ALL | Recruiting | NCT03110354 |
| BETi | RO6870810 | 1 | R/R AML and MDS | Completed | NCT02308761 |
| BETi | OTX015 + azacitidine | 1b/2 | Newly diagnosed AML ineligible for intensive chemotherapy | Withdrawn | NCT02303782 |
| LSD1 inhibitor | GSK2879552 | 1 | R/R AML | Terminated | NCT02177812 |
| LSD1 inhibitor | INCB059872 + ATRA or azacitidine | 1 | R/R AML, newly diagnosed AML | Recruiting | NCT02712905 |
| LSD1 inhibitor | IMG-7289 ± ATRA | 1 | AML and MDS | Completed | NCT02842827 |
| LSD1 inhibitor | Tranylcypromine + ATRA and cytarabine | 1 | AML and MDS | Recruiting | NCT02717884 |
| LSD1 inhibitor | Tranylcypromine + ATRA | 1 | AML and MDS | Active, not recruiting | NCT02273102 |
| PRMT5 inhibitor | GSK3326595 | 1 | AML, MDS, CMML | Recruiting | NCT03614728 |
| Class | Agent | Phase | Target population | Status | Identifier |
|---|---|---|---|---|---|
| HMA | Guadecitabine | 3 | R/R AML | Active, not recruiting | NCT02920008 |
| HMA + DLI | Guadecitabine + DLI | 2 | AML or MDS relapsed post-AlloSCT | Recruiting | NCT02684162 |
| HMA | Guadecitabine | 3 | Newly diagnosed AML ineligible for intensive chemotherapy | Completed | NCT02348489 |
| HMA ± chemotherapy | Guadecitabine ± idarubicin | 2 | AML age ≥70 y | Active, not recruiting | NCT02096055 |
| HMA | Guadecitabine | 2 | AML with 20%-30% blasts or MDS refractory to azacitidine | Completed | NCT02197676 |
| HMA | Guadecitabine | 1/2 | AML, MDS | Completed | NCT01261312 |
| HMA + anti–PD-1 | Guadecitabine + atezolizumab | 1 | AML | Suspended | NCT02892318 |
| HDACi | Entinostat | 2 | AML age ≥60 y | Recruiting | NCT01305499 |
| HDACi + HMA | Entinostat + azacitidine | 1 | AML, MDS, CML | Active, not recruiting | NCT00101179 |
| HDACi + chemotherapy | Panobinostat + cytarabine and daunorubicin | 1 | AML, MDS age ≥60 | Active, not recruiting | NCT01463046 |
| HDACi + chemotherapy | Panobinostat + cytarabine and idarubicin | 1/2 | AML age ≥65 y | Completed | NCT00840346 |
| HDACi + chemotherapy | Panobinostat + cytarabine and idarubicin | 1 | AML age <65 y | Completed | NCT01242774 |
| HDACi + HMA | Panobinostat + azacitidine | 1 | AML <30% blasts, MDL, CMML, | Active, not recruiting | NCT00946647 |
| HDACi + HMA | Panobinostat + decitabine | 1/2 | AML age ≥60, MDS | Completed | NCT00691938 |
| HDACi + HMA | AR-42 + decitabine | 1 | AML | Completed | NCT01798901 |
| HDACi | Belinostat | 2 | AML age ≥60 | Completed | NCT00357032 |
| HDACi + HMA | Mocetinostat + azacitidine | 1/2 | AML, MDS | Completed | NCT00324220 |
| HDACi | Romidepsin | 2 | R/R AML | Completed | NCT00062075 |
| mIDH1 inhibitor | BAY1436032 | 1 | mIDH1 AML | Completed | NCT03127735 |
| mIDH1 inhibitor | Ivosidenib + venetoclax | 1/2 | mIDH1 AML, MDS/MPN | Recruiting | NCT03471260 |
| mIDH1 inhibitor ± HMA, chemotherapy | FT-2102 ± azacitidine or cytarabine | 1/2 | mIDH1 AML, MDS | Recruiting | NCT02719574 |
| mIDH2 inhibitor + HMA | Enasidenib + azacitidine | 2 | R/R mIDH2 AML | Recruiting | NCT03683433 |
| mIDH1, mIDH2 inhibitors + chemotherapy | Ivosidenib or enasidenib + cytarabine and daunorubicin or idarubicin | 1 | Newly diagnosed mIDH1 or mIDH2 AML | Active, not recruiting | NCT02632708 |
| mIDH1, mIDH2 inhibitors + HMA | Ivosidenib or enasidenib + azacitidine | 1/2 | Newly diagnosed mIDH1 or mIDH2 AML ineligible for intensive chemotherapy | Active, not recruiting | NCT02677922 |
| mIDH1 inhibitor + HMA | Ivosidenib or placebo + azacitidine | 3 | Newly diagnosed mIDH1 AML | Recruiting | NCT03173248 |
| BETi | OTX015 (MK-8628) | 1 | AML, ALL, NHL, MM | Completed | NCT01713582 |
| BETi ± HMA | FT-1101 ± azacitidine | 1 | R/R AML | Recruiting | NCT02543879 |
| BETi | GSK525762 | 1 | AML, NHL, MM | Recruiting | NCT01943851 |
| BETi | INCB054329 | 1/2 | AML, MDS/MPN, MM, solid tumor, or lymphoma | Completed | NCT02431260 |
| BETi ± BCL2 inhibitor | ABBV-075 +/− venetoclax | 1 | AML, MM, solid tumors, NHL | Active, not recruiting | NCT02391480 |
| EZH 1/2 inhibitor | DS-3201b | 1 | AML or ALL | Recruiting | NCT03110354 |
| BETi | RO6870810 | 1 | R/R AML and MDS | Completed | NCT02308761 |
| BETi | OTX015 + azacitidine | 1b/2 | Newly diagnosed AML ineligible for intensive chemotherapy | Withdrawn | NCT02303782 |
| LSD1 inhibitor | GSK2879552 | 1 | R/R AML | Terminated | NCT02177812 |
| LSD1 inhibitor | INCB059872 + ATRA or azacitidine | 1 | R/R AML, newly diagnosed AML | Recruiting | NCT02712905 |
| LSD1 inhibitor | IMG-7289 ± ATRA | 1 | AML and MDS | Completed | NCT02842827 |
| LSD1 inhibitor | Tranylcypromine + ATRA and cytarabine | 1 | AML and MDS | Recruiting | NCT02717884 |
| LSD1 inhibitor | Tranylcypromine + ATRA | 1 | AML and MDS | Active, not recruiting | NCT02273102 |
| PRMT5 inhibitor | GSK3326595 | 1 | AML, MDS, CMML | Recruiting | NCT03614728 |
ALL, acute lymphoblastic leukemia; AlloSCT, allogeneic stem cell transplantation; ATRA, all-trans retinoic acid; DLI, donor leukocyte infusion; MM, multiple myeloma; MPN, myeloproliferative neoplasm; NHL, non-Hodgkin lymphoma; PRMT5, protein arginine methyltransferase 5; ±, with or without.
Other investigational therapies
Final targets for epigenetic therapies are enzymes that methylate and demethylate histone lysine and arginine residues. Unlike lysine acetylation, lysine methylation can lead to transcriptional repression or activation, depending on the degree of methylation and residue location.93 Histone lysine methyltransferases and histone lysine demethylases are the major enzymes controlling this dynamic process.
Enhancer of zeste homolog 1 (EZH1), EZH2, and disruptor of telomeric silencing 1-like (DOT1L) are histone lysine methyltransferases under clinical investigation as therapeutic targets. EZH1 and EZH2 catalyze histone 3 lysine 27 (H3K27) trimethylation and are highly expressed in hematopoietic stem cells, where they promote cellular proliferation through S-phase entry and G2-M transition.94 In many malignancies, including AML, overexpression of EZH2 silences genes, such as the tumor suppressor p16.94,95 Interestingly, DOT1L-mediated methylation of H3K79 promotes transcriptional activation instead. Mixed lineage leukemia (MLL), a subset of AML associated with particularly poor prognosis, harbors an oncogenic fusion protein that associates with DOT1L and yields excess lysine methylation.96,97 This association promotes transcription of oncogenes necessary for leukemic transformation in MLL.98
In preclinical studies, EZH2 inhibitors have shown activity against human AML cell lines94 ; however, without concurrent EZH1 inactivation, H3K27 trimethylation persisted.99 In mouse models of AML, EZH1/2 dual inhibition eradicated quiescent leukemic stem cells, a population particularly resistant to conventional chemotherapy.100 A clinical trial investigating an EZH1/2 dual inhibitor in AML and acute lymphocytic leukemia is underway (NCT03110354).
Preclinical studies of DOT1L inhibitors demonstrated activity in vitro and in vivo mouse xenografts, with inhibition of H3K79 methylation and induction of differentiation and apoptosis of MLL+ AML cells.101,102 A phase 1 study of pinometostat (EPZ-5676), a DOT1L inhibitor, in children with R/R MLL demonstrated transient reductions in peripheral or marrow blasts in 40%, although no patients experienced objective response.103 Grade 3/4 AEs included febrile neutropenia, anemia, thrombocytopenia, and respiratory failure. Chromatin immunoprecipitation sequencing demonstrated decreased methylation of MLL target genes at all dose levels tested. A phase 1 study of pinometostat in 51 adults with acute leukemias, including 33 MLL-rearranged, 2 MLL partial-tandem duplication, and 6 MLL wild-type patients, has shown modest clinical activity (NCT01684150).104 In total, 2 patients achieved CR, both of whom harbored MLL gene aberrations.104 The most common grade 3/4 AE was febrile neutropenia (33%).
The first lysine demethylase discovered was lysine-specific demethylase 1 (LSD1), which complexes with the co-REST repressor complex to demethylate H3K4 and H3K9.105 In vitro studies have found that LSD1 inhibitors do not significantly increase lysine methylation, and inhibition of LSD1-mediated transcriptional repression may contribute to its therapeutic effect.106 Finally, emerging evidence supports the ability of LSD1 inhibitors, including tranylcypromine, to induce all-trans retinoic acid susceptibility in nonacute promyelocytic leukemia AML.107 Phase 1 studies of multiple LSD1 inhibitors (INCB059872, IMG-7289, tranylcypromine) in AML are currently underway as monotherapy and combination therapy with all-trans retinoic acid or HMAs (Table 3).
A final potential target of epigenetic modifiers is the family of protein arginine methyltransferases (PRMTs), which catalyze the methylation of arginine residues on histones. Arginine methylation mediates DNA repair, signal transduction, and transcriptional regulation.108 PRMT5, in particular, is required for maintaining the survival and pluripotency of hematopoietic stem cells in the marrow.109 Recent studies have linked aberrant PRMT function to oncogenesis. In MLL+ AML, for example, PRMT1 is recruited as part of an oncogenic transcriptional complex, and knockdown of the enzyme’s expression suppresses leukemic transformation.110 A phase 1 study of the PRMT5 inhibitor GSK3326595 in AML and MDS has recently begun (NCT03614728).
Conclusions
For most AML patients, high-intensity chemotherapy, with or without hematopoietic stem cell transplantation, remains the standard of care, whereas patients ineligible for these therapies have limited treatment options. An increased appreciation for the biologic significance of the leukemic epigenome spurred the development of epigenetic therapies through which meaningful incremental improvements in outcomes have been achieved. HMA and mutant IDH inhibitor monotherapies have addressed an important niche in the treatment of elderly or unfit patients. Unfortunately, preclinical promise for many novel epigenetic therapies have been followed by muted response rates in patients. Thus, investigators have increasingly taken advantage of the ability of epigenetic therapies to modify cellular programming to search for synergistic pairings that may improve efficacy, reduce toxicity, and allow patients to remain ambulatory. Finally, as the repertoire of these novel agents and combinations expand, uncovering biomarkers predictive of response represents an underdeveloped priority.
Acknowledgment
The authors thank Ni-ka Ford, an academic illustrator at the Icahn School of Medicine at Mount Sinai, for exceptional help with creating the figure of this manuscript.
Authorship
Contribution: D.P. researched and wrote the manuscript; R.R. edited the manuscript and provided critical feedback that helped to shape it; and J.M. provided guidance regarding overall direction and wrote and edited the manuscript.
Conflict-of-interest disclosure: R.R. has received consulting fees from Constellation, Incyte, Celgene, Promedior, CTI BioPharma, Jazz Pharmaceuticals, Blueprint, and Stemline Therapeutics and has received research funding from Incyte, Constellation, and Stemline Therapeutics. J.M. has received research funding paid to his institution by CTI BioPharma, Constellation Pharmaceuticals, Roche, Promedior, Janssen, Incyte, Novartis, Merck, Merus, Arog Pharmaceuticals, Kartos Therapeutics, and Celgene and has consulted and acts as a clinical trial and scientific advisory board member for Roche, Constellation Pharmaceuticals, Kartos Therapeutics, Incyte, and Celgene. D.P. declares no competing financial interests.
Correspondence: Darren Pan, Icahn School of Medicine Mount Sinai, 17 East 102nd St, New York, NY 10029; e-mail: darren.pan@mountsinai.org.

