

Key Points
We report rare monoallelic variants of THPO that alter intracellular trafficking and diminish thrombopoietin secretion.
Affected cases have autosomal-dominant thrombocytopenia but no other hematological features.
Introduction
Thrombopoietin (TPO) is a lineage-specific cytokine that is synthesized predominantly in hepatocytes and is an essential regulator of self-renewal of hematopoietic stem cells and their differentiation into platelet-forming megakaryocytes.1,2 The major TPO isoform is a 353-aa peptide containing a highly glycosylated C-terminal domain and an amino-terminal receptor binding domain (RBD) that mediates interaction with the TPO receptor (c-mpl) on hematopoietic cells.3-5 TPO is encoded by the THPO gene, which maps to 3q27.1 and contains up to 6 coding exons.6
Increased TPO production caused by gain-of-function THPO variants results in the myeloproliferative disorder thrombocythemia-1 (Online Mendelian Inheritance in Man #187950), in which the circulating platelet count is elevated, sometimes causing bleeding or thrombosis.7-9 In contrast, biallelic loss-of-function THPO variants result in a severe reduction in the platelet count and multilineage bone marrow failure.10-12 Here, we present genetic and functional evidence that monoallelic loss-of-function THPO variants also result in reduced platelet count but do not affect other circulating blood cells.
Methods
Study subjects were enrolled into the National Institute for Health Research BioResource for Rare Diseases between December 2012 and March 2017 or were referred for analysis with the ThromboGenomics high-throughput diagnostic platform.13 All subjects provided informed written consent in accordance with East of England–Cambridge South research ethics committee approvals (13/EE/0325 and 10/H0304/66).14 Clinical and laboratory data were coded using Human Phenotype Ontology terms.15 DNA preparation, sequencing, and variant calling were performed as described previously.13,14
Using the BeviMed method for genetic association,16 we compared the genotypes at rare nonsynonymous variants of 105 unrelated thrombocytopenia cases in the National Institute for Health Research BioResource for Rare Diseases with those of the other 10 152 unrelated participants who did not have unexplained thrombocytopenia (supplemental Information). Thrombocytopenia cases were defined as patients with a platelet count (PLT) <130 × 109/L or with the Human Phenotype Ontology term “thrombocytopenia” (HP:0001873). Study participants were recalled, and genotype calls were confirmed by polymerase chain reaction DNA amplification and Sanger sequencing (supplemental Information). Blood cell parameters were measured using a Sysmex XN-Series analyzer. Serum TPO levels were measured using an enzyme-linked immunosorbent assay (ELISA; R&D Systems).
To further assess the pathogenicity of the THPO variants, pcDNA3.1+ expression vectors containing N-terminal FLAG-tagged wild-type (WT) THPO or the variant THPO complementary DNAs (cDNAs; GenScript) were transfected into cultured HEK293T cells. Cell supernatants and lysates were collected 24 hours after transfection, and the FLAG-tagged proteins were quantified by ELISA (Cayman Chemical). Transfected cells stained with an antibody against the FLAG epitope were examined by confocal immunofluorescence microscopy (supplemental Information).
Results and discussion
To identify new genes or modes of inheritance for heritable thrombocytopenia, we evaluated whole-genome sequence data for 33 814 genes using the BeviMed method for genetic association. Sixteen genes had a posterior probability of association with thrombocytopenia >0.4, of which 13 have previously been shown to harbor variants causal of thrombocytopenia in multiple pedigrees (Figure 1A). Of the remainder, only THPO harbored rare variants with a high probability of pathogenicity that segregated with thrombocytopenia in family members.
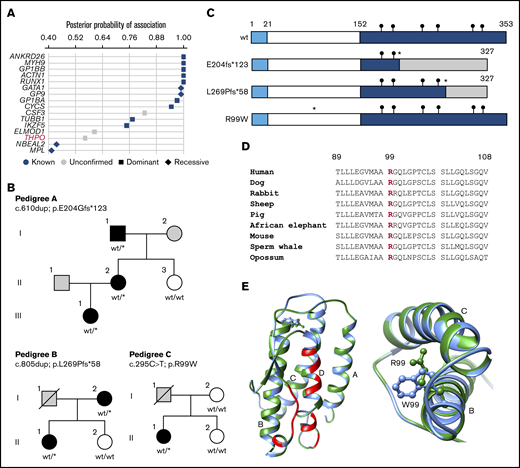
Rare monoallelic variants in THPO are associated with thrombocytopenia. (A) Associations between rare filtered variants across all genes and case/control groups defined by the presence of thrombocytopenia were inferred using the BeviMed method. Genes with a posterior probability of association >0.4 are shown for dominant and recessive inheritance models. Genes previously associated with thrombocytopenia are shown as blue symbols, whereas those with no confirmed association are shown as gray symbols. (B) Pedigree structures of the 3 index cases (A, II.2; B, I.2; and C, II.1) and pedigree members, indicating the presence (black symbols) or absence (white symbols) of thrombocytopenia and cases with unknown PLT (gray symbols). Genotypes confirmed by Sanger sequencing are annotated against canonical transcript ENST00000204615.7 (GRCh37.p13). wt/wt indicates homozygous for the reference allele. wt/* indicates the monoallelic variant. (C) Schematic diagram of the wt and predicted variant TPO proteins with amino acid numbering indicating the signal peptide (1-21), RBD (22-152), and C-terminal domain (153-353). Black circles indicate the position of consensus N-linked glycan sites. The insertion variants p.E204fs*123 and p.L269Pfs*58 (*) predict altered amino acid sequence (shaded region) downstream of the variant site and chain truncation, both after codon 327. The predicted p.R99W variant (*) is in the RBD. (D) Phylogenetic conservation of R99 across representative mammalian species shown in a sequence line up prepared using Clustal Omega. (E) Ribbon diagram of the TPO RBD derived from PDB:1V7M with direct contact regions between TPO and the TPO receptor indicated in red and the 4 helical regions indicated as A, B, C, and D. In the expanded view (left panel) and detailed vertical view (right panel), the wt R99 (green side chain) occurs in the cross-over region between helices A and B in the wt TPO peptide. Substitution of R99 with a W (blue) residue introduces a amphipathic residue with a bulky aromatic side chain into the space between helices B and C.
Rare monoallelic variants in THPO are associated with thrombocytopenia. (A) Associations between rare filtered variants across all genes and case/control groups defined by the presence of thrombocytopenia were inferred using the BeviMed method. Genes with a posterior probability of association >0.4 are shown for dominant and recessive inheritance models. Genes previously associated with thrombocytopenia are shown as blue symbols, whereas those with no confirmed association are shown as gray symbols. (B) Pedigree structures of the 3 index cases (A, II.2; B, I.2; and C, II.1) and pedigree members, indicating the presence (black symbols) or absence (white symbols) of thrombocytopenia and cases with unknown PLT (gray symbols). Genotypes confirmed by Sanger sequencing are annotated against canonical transcript ENST00000204615.7 (GRCh37.p13). wt/wt indicates homozygous for the reference allele. wt/* indicates the monoallelic variant. (C) Schematic diagram of the wt and predicted variant TPO proteins with amino acid numbering indicating the signal peptide (1-21), RBD (22-152), and C-terminal domain (153-353). Black circles indicate the position of consensus N-linked glycan sites. The insertion variants p.E204fs*123 and p.L269Pfs*58 (*) predict altered amino acid sequence (shaded region) downstream of the variant site and chain truncation, both after codon 327. The predicted p.R99W variant (*) is in the RBD. (D) Phylogenetic conservation of R99 across representative mammalian species shown in a sequence line up prepared using Clustal Omega. (E) Ribbon diagram of the TPO RBD derived from PDB:1V7M with direct contact regions between TPO and the TPO receptor indicated in red and the 4 helical regions indicated as A, B, C, and D. In the expanded view (left panel) and detailed vertical view (right panel), the wt R99 (green side chain) occurs in the cross-over region between helices A and B in the wt TPO peptide. Substitution of R99 with a W (blue) residue introduces a amphipathic residue with a bulky aromatic side chain into the space between helices B and C.
Of the 4 heterozygous variants contributing to the high posterior probability of association between THPO and thrombocytopenia, 2 occurred in pedigrees that were available for further phenotype testing (Figure 1B; pedigrees A and C). A third thrombocytopenic case carrying a rare heterozygous THPO variant was identified in the ThromboGenomics collection of cases with undiagnosed bleeding and platelet disorders (Figure 1B; pedigree B). The variants included 2 single-nucleotide insertions (pedigree A, c.610dup; p.E204Gfs*123 and pedigree B, c.805dup; p.L269Pfs*58), both predicting a TPO peptide with an abnormal C terminus with a stop gain after codon 327 causing chain truncation (Figure 1C). The other variant was a missense single-nucleotide variant (pedigree C, c.295C>T; p.R99W) identified in a single case but absent in other available family members (Figure 1B). The substituted TPO R99 residue is highly conserved across eukaryotes (Figure 1D) and is located between α-helices A and B in the TPO RBD, distinct from residues that interface with the TPO receptor.5 Substitution of the charged polar R99 with W has a high CADD score of 33.017 and introduces a bulky amphipathic residue between TPO helical domains B and C (Figure 1E).
The 3 index cases and 3 accessible family members with THPO variants had reduced PLT (median, 120.5 × 109/L; range, 102-143) and increased mean platelet volume (median, 12.4 fL; range, 12.1-14.2) compared with ∼50 000 blood donors from the INTERVAL study (Figure 2A).18 Blood films confirmed thrombocytopenia and showed platelet anisocytosis with some large platelets (Figure 2B). There were no other quantitative blood cell abnormalities (supplemental Information). Mild mucocutaneous bleeding was reported in 4 cases, but there were no other consistent phenotypic features. The median serum TPO concentration in the subjects was 42 pg/mL (range, 17-57; n = 4), which was lower than expected for PLT compared with 35 controls who were healthy or who had low PLT following chemotherapy for hematological malignancy (Figure 2C).

Phenotypic characteristics of monoallelic THPO variants and expression in HEK293T cells. (A) Sex-stratified graphs of PLT and mean platelet volume obtained using a Sysmex XN-Series hematology analyzer from 48 345 blood donors from the INTERVAL study.17 The arrows indicate the platelet parameters of the cases harboring THPO variants. (B) Representative May-Grünwald-Giemsa–stained peripheral blood films from THPO variant cases confirming thrombocytopenia and indicating that some platelets (arrows) were enlarged. Scale bars, 5 μM. (C) Serum TPO concentration plotted against platelet counts in healthy controls or hemato-oncology patients with thrombocytopenia caused by bone marrow suppression after chemotherapy. Corresponding data from 4 cases with THPO variants (A I.1 and II.2, B I.2, C II.1) are also shown. The solid and dotted lines represent the line of best-fit and 95% confidence intervals, respectively, from a nonlinear regression of control data. (D) Effects of THPO variants in HEK293T cells: Flag-tagged WT, E204Gfs*123, L269P*58, or R99W substituted TPO was expressed in HEK293T cells by transient transfection. Secretion of FLAG-tagged TPO in cell supernatant (D) and lysate (E) was measured using an ELISA and normalized against total protein in the cell lysate. Results are mean ± standard error from ≥5 independent experiments. The P values were calculated using 1-way analysis of variance. (F) Representative images from HEK293T cells transfected with WT and variant THPO cDNAs and stained with an antibody against the FLAG epitope (red). Nuclei were stained with 4′,6-diamidino-2-phenylindole (blue). Images were captured using a Leica SP5 II confocal microscope with a 63×/1.4 NA lens and analyzed with ImageJ/Fiji. All images are shown with the same intensity range. (G) Quantification of FLAG-tagged protein by mean fluorescence intensity in representative cells (n = 5).
Phenotypic characteristics of monoallelic THPO variants and expression in HEK293T cells. (A) Sex-stratified graphs of PLT and mean platelet volume obtained using a Sysmex XN-Series hematology analyzer from 48 345 blood donors from the INTERVAL study.17 The arrows indicate the platelet parameters of the cases harboring THPO variants. (B) Representative May-Grünwald-Giemsa–stained peripheral blood films from THPO variant cases confirming thrombocytopenia and indicating that some platelets (arrows) were enlarged. Scale bars, 5 μM. (C) Serum TPO concentration plotted against platelet counts in healthy controls or hemato-oncology patients with thrombocytopenia caused by bone marrow suppression after chemotherapy. Corresponding data from 4 cases with THPO variants (A I.1 and II.2, B I.2, C II.1) are also shown. The solid and dotted lines represent the line of best-fit and 95% confidence intervals, respectively, from a nonlinear regression of control data. (D) Effects of THPO variants in HEK293T cells: Flag-tagged WT, E204Gfs*123, L269P*58, or R99W substituted TPO was expressed in HEK293T cells by transient transfection. Secretion of FLAG-tagged TPO in cell supernatant (D) and lysate (E) was measured using an ELISA and normalized against total protein in the cell lysate. Results are mean ± standard error from ≥5 independent experiments. The P values were calculated using 1-way analysis of variance. (F) Representative images from HEK293T cells transfected with WT and variant THPO cDNAs and stained with an antibody against the FLAG epitope (red). Nuclei were stained with 4′,6-diamidino-2-phenylindole (blue). Images were captured using a Leica SP5 II confocal microscope with a 63×/1.4 NA lens and analyzed with ImageJ/Fiji. All images are shown with the same intensity range. (G) Quantification of FLAG-tagged protein by mean fluorescence intensity in representative cells (n = 5).
To further assess the pathogenicity of the THPO variants, FLAG-tagged WT or variant THPO cDNAs were transfected into HEK293T cells. FLAG-tagged TPO (FLAG-TPO) concentrations measured by immunoassay were lower in the culture supernatants of the variant-transfected cells compared with WT-transfected controls (Figure 2D). These findings strongly support pathogenicity of the observed variants and indicate that thrombocytopenia in the cases results from diminished secretion of TPO.
In cell lysates, the FLAG-TPO concentration in E204Gfs*123 cells was markedly higher than in WT controls (Figure 2E). This was confirmed by confocal immunofluorescence microscopy (Figure 2F-G) and suggests that secretion failure occurs because of mistrafficking and cytoplasmic accumulation of the substituted TPO. Lysates from the L269Pfs*58 and R99W cells contained similar overall concentrations of FLAG-TPO as WT control cells (Figure 2E). However, these substituted TPO peptides showed greater perinuclear localization and less punctate cytoplasmic expression compared with WT (Figure 2F-G). This appearance also suggests mistrafficking, although, with these variants, the substituted TPO proteins appear to accumulate in the Golgi and undergo intracellular degradation rather than accumulating in the cytoplasm. Although these observations provide an attractive mechanism for the reduction in TPO secretion, it is premature to extrapolate these findings in heterologous cells to make inferences about endogenous TPO production in the study cases.
It has been shown previously that mutagenesis of glycosylated residues in the TPO C terminus or chain truncation disrupt TPO secretion,3,19 suggesting that this domain has an essential function in ensuring appropriate trafficking to the secretory compartment. Our findings with the E204Gfs*123 and L269Pfs*58 variants are consistent with this hypothesis, because these variants result in loss of TPO C-terminal domain glycosylated residues (Figure 1C). The monoallelic R99W variant is not predicted to truncate the TPO C-terminal domain; instead, it is likely to alter TPO tertiary structure, potentially disrupting trafficking. Pathogenicity of the R99W variant is further supported by observations from other investigators that biallelic R99W variants caused trilineage bone marrow failure in 2 pedigrees in which affected cases had undetectable serum TPO,11 suggesting defective TPO secretion. It is also noteworthy that thrombocytopenia is sometimes part of the complex phenotype associated with microdeletions of the 3q26.33-3q27.2 region that includes THPO.20-22 A high-impact monoallelic THPO variant predicting p.R31* has also been reported previously in 2 pedigrees with isolated thrombocytopenia, although it was not characterized functionally.23
Heritable thrombocytopenia is a heterogeneous group of disorders associated with variants in a rapidly expanding number of genes, including some associated with severe bleeding, leukemia susceptibility, or nonhematological phenotypes.24,25 Our findings extend the repertoire of this group of disorders and establish that THPO-related thrombocytopenia is caused by monoallelic loss-of-function variants in THPO that result in reduced TPO secretion. Our data suggest that monoallelic THPO variants confer a mild clinical phenotype with abnormalities confined to the platelet lineage.
The whole-genome sequencing data reported in this article have been deposited in the European Genome-phenome Archive (EGAD00001004519). The frameshift variants have been deposited in ClinVar under accession numbers SCV000899945.1 and SCV000899830.1.
Acknowledgments
The authors thank National Institute for Health Research (NIHR) BioResource volunteers for participation and gratefully acknowledge the contributions of NIHR BioResource centres, NHS Trusts, and staff. They also thank the NIHR and NHS Blood and Transplant.
This work was supported by the NIHR through the Biomedical Research Centre at University Hospitals Bristol NHS Foundation Trust and the University of Bristol. N.C. was supported by the Elizabeth Blackwell Institute for Health Research, University of Bristol, and the Wellcome Trust Institutional Strategic Support Fund. K.B. is funded through a GW4 Wellcome Trust fellowship. J.C. is funded through a Medical Research Council Clinical Research Training Fellowship. S.K.W. is funded through an NIHR Clinical Lectureship.
The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health and Social Care.
Authorship
Contribution: N.C., M.R.A., L.F., K.B., and D.A. performed laboratory tests; J.C. and S.K.W. collected phenotype data; K.D. performed high-throughput sequencing and variant analysis; A.D.M. and N.C. wrote the manuscript; and D.G. and E.T. performed statistical genetic analyses and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
A complete list of the members of the NIHR BioResource–Rare Diseases Consortium appears in the online appendix and can be accessed at https://bioresiurce.nihr.ac.uk/researchers/researchers/acknowledgement/.
Correspondence: Naomi Cornish, University of Bristol, Research Floor 7, Bristol Royal Infirmary, Bristol BS2 8HW, United Kingdom; e-mail: nc17663@bristol.ac.uk.

