

TO THE EDITOR:
The frequency of germline RUNX1 variants in an unselected acute myeloid leukemia (AML) population is poorly defined and likely underestimated. The recent study by Simon et al1 is particularly important as a first attempt to define this underlying frequency. Because RUNX1 is part of most next-generation sequencing panels performed on leukemic samples, germline variants are invariably found, highlighted by this and other studies.1-4 Human and medical geneticists, genetic counselors, molecular pathologists, hematopathologists, and hematologists are particularly likely to encounter patients with germline RUNX1 variants and may benefit from guidance on how to interpret these variants and their clinical implications.
In the Simon et al1 study, 10.7% (44/430) of AML patients had a somatic or germline RUNX1 variant. Germline variants represented 27.3% (12/44) of RUNX1 variants, suggesting a 2.8% frequency of germline RUNX1 variants in an unselected AML population. However, it was not clearly delineated whether the identified germline variants were all disease causing (ie, pathogenic or likely pathogenic), although the term “mutation” implies pathogenicity. Inconsistent usage of “variant” and “mutation” can lead to miscommunication of scientific findings, as well as clinical testing results: “mutation” refers to pathogenic/likely pathogenic variations that are deleterious and found less frequently in a population or are nongermline changes in a tumor cell (somatic mutations) that are predictive/therapeutic, diagnostic, or prognostic biomarkers (Table 1).5
Nomenclature of variants and likelihood of being disease causing
| Nomenclature | Definition | Likelihood of being disease causing9,19-21 ,* |
|---|---|---|
| Mutation | “Mutation” is used for germline variations that are pathogenic and found less frequently in a population or are nongermline changes in a tumor cell (somatic mutations).5 | Should only be used when there is clear evidence for pathogenicity |
| Variant | An alteration in the most common DNA nucleotide sequence. The term “variant” can be used to describe an alteration that may be benign, pathogenic, or of unknown significance. The term “variant” is increasingly being used in place of the term “mutation.”22 | Variant is further classified in a 5-tier system: benign, likely benign, uncertain significance, likely pathogenic, pathogenic |
| Benign variant | This variant does not cause disease. | <0.1% |
| Likely benign variant | This variant is not expected to cause disease. Additional evidence may confirm this assertion of benign, but there is a small chance that new evidence may demonstrate that this variant does have clinical significance. | Between 0.1% and 10% |
| Variant of uncertain significance | There is insufficient evidence to put this variant into a benign or pathogenic category. Further evidence, such as population, segregation, or functional data, may up- or downgrade this variant. This variant is not clinically actionable. | Between 10% and 90% |
| Likely pathogenic variant | This variant is expected to cause disease. Additional evidence may confirm this assertion of pathogenicity, but there is a small chance that new evidence may demonstrate that this variant does not have clinical significance. | Between 90% and 99% |
| Pathogenic variant | This variant does cause disease. | >99% |
| Nomenclature | Definition | Likelihood of being disease causing9,19-21 ,* |
|---|---|---|
| Mutation | “Mutation” is used for germline variations that are pathogenic and found less frequently in a population or are nongermline changes in a tumor cell (somatic mutations).5 | Should only be used when there is clear evidence for pathogenicity |
| Variant | An alteration in the most common DNA nucleotide sequence. The term “variant” can be used to describe an alteration that may be benign, pathogenic, or of unknown significance. The term “variant” is increasingly being used in place of the term “mutation.”22 | Variant is further classified in a 5-tier system: benign, likely benign, uncertain significance, likely pathogenic, pathogenic |
| Benign variant | This variant does not cause disease. | <0.1% |
| Likely benign variant | This variant is not expected to cause disease. Additional evidence may confirm this assertion of benign, but there is a small chance that new evidence may demonstrate that this variant does have clinical significance. | Between 0.1% and 10% |
| Variant of uncertain significance | There is insufficient evidence to put this variant into a benign or pathogenic category. Further evidence, such as population, segregation, or functional data, may up- or downgrade this variant. This variant is not clinically actionable. | Between 10% and 90% |
| Likely pathogenic variant | This variant is expected to cause disease. Additional evidence may confirm this assertion of pathogenicity, but there is a small chance that new evidence may demonstrate that this variant does not have clinical significance. | Between 90% and 99% |
| Pathogenic variant | This variant does cause disease. | >99% |
Most variants do not have data to support a quantitative assignment of variant certainty to any of the 5 categories given the heterogeneous nature of most diseases.
Germline variant classification is performed using 5 ranks of pathogenicity: pathogenic, likely pathogenic, variant of uncertain significance, likely benign, and benign. Variants of uncertain significance, as well as likely benign and benign variants, should not be attributed to disease causality (Table 1). Accurate variant classification is critically important for attribution of pathogenicity of the identified variants and their actionability, because the identification of a deleterious germline variant has clinical implications that extend far beyond the treatment of the diagnosed individual.
In response to interlaboratory curation differences, the Clinical Genome Resource (ClinGen) has launched Variant Curation Expert Panels (VCEPs) to develop gene- or disease-specific American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) criteria.6 The Myeloid Malignancy (MM)-VCEP was formed in 2018 and published RUNX1-specific ACMG/AMP criteria in 2019.7,8 Given our familiarity with the RUNX1 variant curation rules, we have reviewed the variants described in the Simon et al1 study and found that only 7 of the 12 germline variants meet the criteria for pathogenic/likely pathogenic classification (Table 2). Thus, the actual yield of deleterious germline RUNX1 variants is 16% (7/44) of all RUNX1 variants and 1.6% (7/430) of all AML patients. Other than early truncating variants leading to non-sense–mediated decay, most causative RUNX1 variants are dependent on a variety of pathogenic evidence. In the case of RUNX1, this is usually a combination of computational and predictive, functional, population, and segregation data in a Bayesian framework.7,9
Comparison of RUNX1 variant curation between the study by Simon et al and the MM-VCEP
| ID* | Variant cDNA/protein* | Described in MDS/AML* | Described in RUNX1 FPD* | Functional impact on RUNX1* | MM-VCEP ACMG/AMP criteria code* | MM-VCEP RUNX1-specific criteria† | Further explanation of criteria† | MM-VCEP classification† |
|---|---|---|---|---|---|---|---|---|
| 1 | c.44_45delAG/p.Q15fsX | — | — | Truncating | PVS1_moderate, PS4_supporting, PM2 | PS4_supporting, PM2 | PVS1 cannot be used for early truncating variants only affecting RUNX1 isoform C. | VUS |
| 2 | c.179C>T/p.A60V | Carnicer et al23 | Lorente24 | — | BS1 | BS1, BS3 | This variant meets the calculated BS1 threshold (Latino subpopulation) and BS3 (normal transactivation and normal DNA binding/subcellular localization).25 The presence of the variant in patients with a RUNX1 phenotype is not sufficient to call a variant PATH, in particular not if the variant is present in gnomAD at a MAF incompatible with disease prevalence. | BEN |
| 3+4‡ | c.421T>G/p.S141A | — | RUNX1db | Normal transactivation26 | PS4_supporting, PP3, BS3_supporting | PM1_supporting, PP3 | Variant not present in RUNX1db. Although there is no effect on heterodimerization ability with CBF,26 data from an additional secondary assay or transactivation assay are missing; this does not permit application of any BS3 strength level. PS4 cannot be applied (2 alleles in gnomAD). | VUS |
| 5 | c.427G>T/p.E143X | — | — | Truncating | PVS1, PS4_supporting, PM2 | PVS1, PS4_supporting, PM2 | PATH | |
| 6 | c.454_456insA/p.K152fsX | Ernst et al27 | — | Truncating | PVS1, PS4_supporting, PM2 | PVS1, PS4_supporting, PM2 | Variant nomenclature does not conform with HGVS recommendations for sequence variants. We assume this variant is not present in gnomAD (PM2) and leads to NMD (PVS1). | PATH |
| 7 | c.496C>G/p.R166G | Imai et al28 | — | LOF/dominant negative28 | PS4_supporting, PM2, PM5, PP3 | PS4_supporting, PM1, PM2, PM5, PP3 | R166Q has been curated by the MM-VCEP as PATH. | LPATH |
| 8 | c.496C>T/p.R166X | Preudhomme et al29 | Bluteau et al30 | Truncating | PVS1, PS4, PM2, PP1 | PVS1, PS4, PM2, PP1_strong | PATH | |
| 9+10 | c.610C>T/p.R204X | Osato et al31 | Song et al32 | LOF31 | PVS1, PS4, PM2, PP1 | PVS1, PS4, PM2, PP1_strong | PATH | |
| 11 | c.619C>T/p.R207W | You et al33 | — | — | PS4_supporting, PM2, PP3 | PS4_moderate, PM2, PP3 | In silico prediction alone (ie, in this case pathogenic predictions by using SIFT, Polyphen, VEST, CHASM, and REVEL) is only supporting evidence and insufficient to classify a variant as PATH. | VUS |
| 12 | c.1243_1244insC/p.Q415fsX | — | — | Elongated RUNX1 isoform | PVS1_strong, PS4_supporting, PM2 | PVS1_strong, PS4_supporting, PM2 | LPATH |
| ID* | Variant cDNA/protein* | Described in MDS/AML* | Described in RUNX1 FPD* | Functional impact on RUNX1* | MM-VCEP ACMG/AMP criteria code* | MM-VCEP RUNX1-specific criteria† | Further explanation of criteria† | MM-VCEP classification† |
|---|---|---|---|---|---|---|---|---|
| 1 | c.44_45delAG/p.Q15fsX | — | — | Truncating | PVS1_moderate, PS4_supporting, PM2 | PS4_supporting, PM2 | PVS1 cannot be used for early truncating variants only affecting RUNX1 isoform C. | VUS |
| 2 | c.179C>T/p.A60V | Carnicer et al23 | Lorente24 | — | BS1 | BS1, BS3 | This variant meets the calculated BS1 threshold (Latino subpopulation) and BS3 (normal transactivation and normal DNA binding/subcellular localization).25 The presence of the variant in patients with a RUNX1 phenotype is not sufficient to call a variant PATH, in particular not if the variant is present in gnomAD at a MAF incompatible with disease prevalence. | BEN |
| 3+4‡ | c.421T>G/p.S141A | — | RUNX1db | Normal transactivation26 | PS4_supporting, PP3, BS3_supporting | PM1_supporting, PP3 | Variant not present in RUNX1db. Although there is no effect on heterodimerization ability with CBF,26 data from an additional secondary assay or transactivation assay are missing; this does not permit application of any BS3 strength level. PS4 cannot be applied (2 alleles in gnomAD). | VUS |
| 5 | c.427G>T/p.E143X | — | — | Truncating | PVS1, PS4_supporting, PM2 | PVS1, PS4_supporting, PM2 | PATH | |
| 6 | c.454_456insA/p.K152fsX | Ernst et al27 | — | Truncating | PVS1, PS4_supporting, PM2 | PVS1, PS4_supporting, PM2 | Variant nomenclature does not conform with HGVS recommendations for sequence variants. We assume this variant is not present in gnomAD (PM2) and leads to NMD (PVS1). | PATH |
| 7 | c.496C>G/p.R166G | Imai et al28 | — | LOF/dominant negative28 | PS4_supporting, PM2, PM5, PP3 | PS4_supporting, PM1, PM2, PM5, PP3 | R166Q has been curated by the MM-VCEP as PATH. | LPATH |
| 8 | c.496C>T/p.R166X | Preudhomme et al29 | Bluteau et al30 | Truncating | PVS1, PS4, PM2, PP1 | PVS1, PS4, PM2, PP1_strong | PATH | |
| 9+10 | c.610C>T/p.R204X | Osato et al31 | Song et al32 | LOF31 | PVS1, PS4, PM2, PP1 | PVS1, PS4, PM2, PP1_strong | PATH | |
| 11 | c.619C>T/p.R207W | You et al33 | — | — | PS4_supporting, PM2, PP3 | PS4_moderate, PM2, PP3 | In silico prediction alone (ie, in this case pathogenic predictions by using SIFT, Polyphen, VEST, CHASM, and REVEL) is only supporting evidence and insufficient to classify a variant as PATH. | VUS |
| 12 | c.1243_1244insC/p.Q415fsX | — | — | Elongated RUNX1 isoform | PVS1_strong, PS4_supporting, PM2 | PVS1_strong, PS4_supporting, PM2 | LPATH |
All variants are annotated using RefSeq ID NM_001754.4. PS4 is applied assuming that the variants in the Simon et al1 study are germline variants. Germline status should be confirmed in DNA derived from cultured skin fibroblasts, cultured bone marrow mesenchymal stromal cells, or hair roots.
—, no data; BEN, benign; cDNA, complementary DNA; FPD, familial platelet disorder; gnomAD, Genome Aggregation Database; HGVS, Human Genome Variation Society; LOF, loss of function; LPATH, likely pathogenic; MAF, minor allele frequency; NMD, non-sense–mediated decay, PATH, pathogenic, VUS, variant of unknown significance.
From the Simon et al1 study.
MM-VCEP assessment.
Patients are related.
With regard to RUNX1 variant curation in the Simon et al1 study as an example, we would like to highlight the following points. (1) Three major RUNX1 isoforms (A, B, and C) are expressed by the use of 2 promoters and alternative splicing. Isoform function, biological relevance, and expression differ in hematopoietic tissue,10,11 which makes PVS1 not applicable for N-terminal truncating variants affecting only isoform C.7,12 (2) Different strength levels of pathogenic functional evidence (PS3) are based on decreased or enhanced transactivation activity, with or without a secondary assay showing decreased DNA binding affinity, diminished heterodimerization ability with CBFb, abnormal cellular localization, reduced colony-forming potential, or abnormal function of mutant RUNX1 in vivo. Of note, other functional assays, such as interaction of RUNX1 with MLL, are not valid secondary assays for RUNX1 function.13,14 (3) The presence of a RUNX1 germline variant in a proband with hereditary myelodysplastic syndrome (MDS)/acute leukemia, even with the typical phenotype including lifelong thrombocytopenia and platelet dysfunction, does not justify a pathogenic classification, but it is always dependent on a combination of additional functional, cosegregation, predictive, or population data. More information regarding the application of RUNX1-specific ACMG/Association for Molecular Pathology criteria in the classification of variants identified in this study is shown in Table 2.
As of 30 June 2020, 591 RUNX1 variants have been reported in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/). Many germline disease-causing RUNX1 variants are unique to individuals or families; thus, detailed annotation is not always available for reference when a new RUNX1 variant is identified.15 Only 21% of RUNX1 variants are clinically significant (pathogenic/likely pathogenic), whereas the majority (79%) are benign/likely benign or variants of uncertain significance, which are not clinically actionable (Figure 1). It is worth mentioning that 50% of RUNX1 variants are variants of uncertain significance that warrant more collaborative efforts for the scientific community to up- or downgrade them based on new evidence, such as observation in multiple probands, segregation with disease, or functional impact of the variant or absence in affected individuals, nonsegregation with disease, or no effects on protein function.
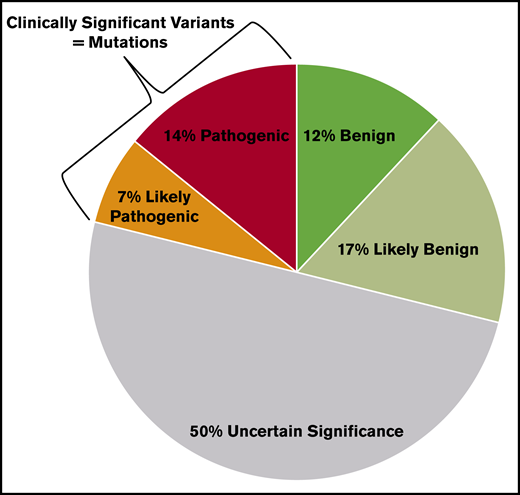
RUNX1 variants in ClinVar and their clinical significance. All 591 RUNX1 variants deposited in ClinVar as of 30 June 2020 and their clinical significance based on the 5-tier system for germline variants (benign, likely benign, variant of uncertain significance, likely pathogenic, pathogenic) are shown in the pie chart. Only 21% of the RUNX1 variants are clinically actionable (ie, likely pathogenic and pathogenic).
RUNX1 variants in ClinVar and their clinical significance. All 591 RUNX1 variants deposited in ClinVar as of 30 June 2020 and their clinical significance based on the 5-tier system for germline variants (benign, likely benign, variant of uncertain significance, likely pathogenic, pathogenic) are shown in the pie chart. Only 21% of the RUNX1 variants are clinically actionable (ie, likely pathogenic and pathogenic).
Phenotypic criteria have been proposed by the ClinGen MM-VCEP, and they can be helpful in the determination of RUNX1 variant pathogenicity, because a high penetrance, with regard to thrombocytopenia and/or underlying platelet dysfunction, is typically recognized, and patients display ≥1 of the following features7 : mild to moderate thrombocytopenia with normal platelet size and volume in the absence of other causative factors; platelet ultrastructural and/or functional defects; and diagnosis of a hematologic malignancy, most commonly affecting the myeloid lineage (causing AML or MDS) and less frequently involving the lymphoid lineage and manifesting as T-cell acute lymphoblastic leukemia or others.
The following example highlights the importance of variant annotation for management decisions. A 56-year-old female with a diagnosis of MDS and a family history of hematologic malignancies was identified to have a germline RUNX1 c.167T>C (p.Leu56Ser) variant and was counseled that this RUNX1 variant was disease causing. Family members were tested for this variant to determine who lacked the variant and, thus, could be an appropriate stem cell transplant donor for the index patient and who in the family carries the variant and should receive surveillance on research protocols for RUNX1-associated familial platelet disorder with myeloid malignancy. Importantly, upon further review at the time of a second opinion, the RUNX1 c.167T>C (p.Leu56Ser) variant was reclassified to be a benign germline variant and a “red herring” in the evaluation of this family.
Patients with chronic otherwise unexplained thrombocytopenia, platelet ultrastructural and/or functional defects, and/or AML, MDS, or T-cell acute lymphoblastic leukemia should undergo genetic testing whenever there is a positive family history for a RUNX1 phenotype and when the patient has been diagnosed at a young age or a RUNX1 variant has been identified upon molecular testing of the leukemic clone. Germline material for testing should represent tissues that are not contaminated with blood/circulating blasts, such as cultured skin fibroblasts, which are the gold standard. Upon confirmation of a germline disease-causing RUNX1 variant, additional family members can be tested and followed-up long-term, including a baseline bone marrow biopsy with cytogenetic/molecular analysis and additional biopsies at the time of any significant/persistent change in blood counts. Most importantly, a family member with the RUNX1 variant should not be considered as a related stem cell donor, which makes recognition of the underlying germline syndrome paramount.16-18
Our clinical example and the variant interpretation by Simon et al1 highlight how easily variants can be misclassified when criteria are not applied correctly, too much weight is put on the observation of the variant in affected probands, or the criteria are not combined correctly to reach the level of clinical significance (ie, disease causing). The accuracy of RUNX1 variant classification and interpretation is of great importance for treatment and follow-up of affected patients, related donor selection, and counseling of family members. Therefore, we emphasize that MM-VCEP RUNX1-specific rules, as the most accurate standards of germline RUNX1 variant classification, should be applied in clinical and research settings.7,8
Data sharing requests should be sent to Simone Feurstein (feurstein@uchicago.edu).
Acknowledgments:
This work was supported by the National Human Genome Research Institute National Institutes of Health (grants U41HG009649 and U41HG009650) and a 2018 National Institutes of Health/National Cancer Institute Leukemia SPORE DRP award (P50CA100632-16, project 00007529) (C.D.D.). The results provided in this publication were generated by the American Society of Hematology in collaboration with Baylor College of Medicine and the University of North Carolina, National Institutes of Health–funded ClinGen grant award recipients.
This commentary was written on behalf of the collaborative group of the American Society of Hematology - Clinical Genome Resource MM-VCEP, which is cochaired by Lucy A. Godley (University of Chicago) and David Wu (University of Washington). A complete list of MM-VCEP members is available at: https://clinicalgenome.org/affiliation/50034/#heading_membership.
Contribution: S.F. reviewed and classified the RUNX1 variants and explained the use of RUNX1-specific criteria; L.Z. worked on the nomenclature of variants; C.D.D. focused on the clinical impact of germline RUNX1 variants; and all authors wrote and edited the manuscript.
Conflict-of-interest disclosure: L.Z. has received honoraria from Future Technology Research LLC, BGI, and Illumina and honoraria and travel and accommodation expenses from Roche Diagnostics Asia Pacific; family members hold leadership positions and ownership interests in the Shanghai Genome Center. The remaining authors declare no competing financial interests.
Correspondence: Simone Feurstein, The University of Chicago, KCBD 7123A, 900 East 57th St, Chicago, IL 60637; e-mail: feurstein@uchicago.edu.

