

Key Points
The FXI gene arose from a duplication of the gene for prekallikrein during mammalian evolution.
Changes in FXI over time allow it to function independently of the kallikrein-kinin system in placental mammals.

Abstract
Factor XI (FXI) is the zymogen of a plasma protease (FXIa) that contributes to hemostasis by activating factor IX (FIX). In the original cascade model of coagulation, FXI is converted to FXIa by factor XIIa (FXIIa), a component, along with prekallikrein and high-molecular-weight kininogen (HK), of the plasma kallikrein-kinin system (KKS). More recent coagulation models emphasize thrombin as a FXI activator, bypassing the need for FXIIa and the KKS. We took an evolutionary approach to better understand the relationship of FXI to the KKS and thrombin generation. BLAST searches were conducted for FXI, FXII, prekallikrein, and HK using genomes for multiple vertebrate species. The analysis shows the KKS appeared in lobe-finned fish, the ancestors of all land vertebrates. FXI arose later from a duplication of the prekallikrein gene early in mammalian evolution. Features of FXI that facilitate efficient FIX activation are present in all living mammals, including primitive egg-laying monotremes, and may represent enhancement of FIX-activating activity inherent in prekallikrein. FXI activation by thrombin is a more recent acquisition, appearing in placental mammals. These findings suggest FXI activation by FXIIa may be more important to hemostasis in primitive mammals than in placental mammals. FXI activation by thrombin places FXI partially under control of the vitamin K-dependent coagulation mechanism, reducing the importance of the KKS in blood coagulation. This would explain why humans with FXI deficiency have a bleeding abnormality, whereas those lacking components of the KKS do not.
Introduction
Advances in nucleic acid sequencing have made whole genomes available for many organisms, facilitating reconstruction of the natural histories of complex processes such as blood coagulation. Pioneering work by Russel Doolittle and colleagues showed that all vertebrate organisms possess a system for thrombin generation based on vitamin K-dependent proteases and their cofactors that is triggered when factor VII/VIIa (FVII/FVIIa) binds to the membrane protein tissue factor at an injury site (Figure 1A).1-5 In some vertebrates, thrombin generation is also initiated by a distinct process called contact activation (Figure 1B).6,7 When blood is exposed to a variety of biologic or nonbiologic “surfaces,” proteins of the kallikrein-kinin system (KKS) become activated. The KKS consists of the protease zymogens prekallikrein (PK) and factor XII (FXII) and the cofactor high-molecular-weight kininogen (HK).6-9 On surfaces, PK and FXII are converted to the active proteases plasma kallikrein (PKa) and FXIIa. FXIIa converts factor XI (FXI) to FXIa, which engages the thrombin generation mechanism through factor IX (FIX) (Figure 1C).10-13 This sequence of reactions, referred to as the intrinsic pathway, initiates clotting in the activated partial thromboplastin time (aPTT) assay used in clinical practice.
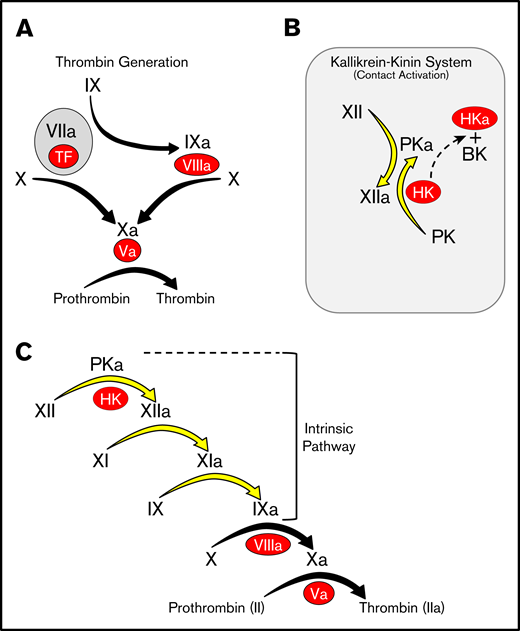
Models of thrombin generation. For all panels, plasma proteases are indicated in black lettering, with the activated forms indicated by a lower case “a.” Cofactors are indicated in red ovals. (A) Thrombin generation. Major protease activation reactions during thrombin generation are indicated by black arrows. Thrombin generation at a wound is typically initiated by a complex formed between the plasma protease factor VIIa and the cofactor TF. (B) The kallikrein-kinin system (KKS). On a surface (represented by the gray rectangle), factor XII (FXII), and PK undergo reciprocal activation to FXIIa and PKa. PKa cleaves HK, releasing BK. (C) The cascade-waterfall model of thrombin generation. In this model, the process is initiated by activation of FXII through the reactions shown in panel B. FXIIa then converts FXI to FXIa, which then activates FIX. The series of reactions indicated by the yellow arrows is referred to as the intrinsic pathway. Image adapted from Gailani and Gruber.12 BK, bradykinin; TF, tissue factor.
Models of thrombin generation. For all panels, plasma proteases are indicated in black lettering, with the activated forms indicated by a lower case “a.” Cofactors are indicated in red ovals. (A) Thrombin generation. Major protease activation reactions during thrombin generation are indicated by black arrows. Thrombin generation at a wound is typically initiated by a complex formed between the plasma protease factor VIIa and the cofactor TF. (B) The kallikrein-kinin system (KKS). On a surface (represented by the gray rectangle), factor XII (FXII), and PK undergo reciprocal activation to FXIIa and PKa. PKa cleaves HK, releasing BK. (C) The cascade-waterfall model of thrombin generation. In this model, the process is initiated by activation of FXII through the reactions shown in panel B. FXIIa then converts FXI to FXIa, which then activates FIX. The series of reactions indicated by the yellow arrows is referred to as the intrinsic pathway. Image adapted from Gailani and Gruber.12 BK, bradykinin; TF, tissue factor.
Individuals lacking KKS components do not have obvious bleeding abnormalities, suggesting thrombin generation through the intrinsic pathway is not required for hemostasis.6-8 However, deficiency of FXI, a major FXIIa substrate, does compromise hemostasis,14,15 indicating a more complex relationship with thrombin generation than is suggested by the cascade/waterfall model (Figure 1C). We took an evolutionary approach to better understand the relationships between FXI, the KKS, and thrombin generation. In 2008, we surveyed the limited collection of available vertebrate genomes and proposed that the KKS arose in early terrestrial vertebrates, with FXI appearing later in marsupial mammals.16 Here we take advantage of genomic data compiled over the past decade to correct and refine our hypothesis. The results show that the KKS arose much earlier than previously suspected, in lobe-finned fish, the ancestors of terrestrial vertebrates. FXI also appeared earlier in mammalian evolution than originally proposed and has undergone changes over time that have facilitated its transitions from KKS protease to an integrated component of the thrombin generation mechanism in placental mammals.
Methods
Materials
Normal human plasma (Precision Biologics); human plasmas lacking PK, FXII, HK, or FXI (George King Biomed); and plasma from a false killer whale (Vancouver Aquarium) were anticoagulated with sodium citrate. Human FXII, FXIIa, PK, HK, and FIX were from Enzyme Research Laboratories and FXI and α-thrombin were from Haematologic Industries. Polyphosphate (100-300 phosphate units in length) not saturated with calcium, was provided by James Morrissey, University of Michigan.
Genome analyses
Genomes of organisms listed in supplemental Figure 1 were surveyed with BLAST searches using human protein sequences for PK, FXI, FXII, HK, and pro-hepatocyte growth factor activator (pro-HGFA) against the National Center for Biotechnology Information nucleotide and protein databases of whole genomic and transcriptomic sequences, and single read archive data from next-generation sequencing. Results were examined by techniques described in supplemental Figure 116 and by visual inspection.
Recombinant proteins
pJVCMV expression constructs for human FXI wild type (FXI-WT) and FXI with the A3 domain replaced with human PK-A3 sequence (FXI-PKA3) are described.17,18 FXI-A3 was also replaced with duck-billed platypus FXI-A3 (FXI/PlatXIA3) or PK-A3 (FXI/PlatPKA3; supplemental Figure 2). Lys367-Pro368 in FXI-WT was replaced with Arg-Ser to generate FXI-PlatRSR (supplemental Figure 2). FXIs were expressed in HEK293 fibroblasts and purified by anti-FXI immunoglobulin G (IgG) affinity chromatography.18 FXIa was prepared by incubating FXI (1.0-2.0 μM) in 20 mM Tris-HCl, 100 mM NaCl, pH 7.4 with FXIIa (5 nM). Human FXII19,20 and pro-HGFA21 complementary DNAs (cDNAs) in pJVCMV modified with C-terminal hemagglutinin tags were expressed in HEK293 cells and purified and activated as shown in supplemental Figure 3.
FIX and FXI activation.
FIX (200 nM) in 20 mM Tris-HCl, 100 mM NaCl, 5 mM CaCl2, pH 7.4 was incubated at 37°C with vehicle or FXIa (2 nM). FXI (200 nM) in 20 mM Tris-HCl, 100 mM NaCl, 4 μM polyphosphate, pH 7.4 was incubated at 37°C with vehicle, FXIIa (40 nM), or α-thrombin (140 nM). At various times, aliquots were removed into reducing sodium dodecyl sulfate (SDS) sample buffer, size fractionated on 10% polyacrylamide-SDS gels, and stained with Coomassie Blue.
PK activation.
PK (200 nM) in 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid pH 7.4, 100 mM NaCl, 1 mg/mL polyethylene glycol-8000, 10 μg/mL dextran sulfate (500 KDa) was incubated (37°C) with 25 nM FXIIa or HGFA or vehicle. Aliquots were removed at various times into reducing SDS-sample buffer, fractionated on 10%-polyacrylamide-SDS gels, and transferred to nitrocellulose. Western blots were developed with horseradish peroxidase (HRP)-conjugated anti-human PK IgG. Samples were tested for PKa activity by incubating with 0.5 mM S-2302 and measuring ΔOD405 nm.
aPTT assay
Human normal or factor-deficient plasma (30 μL) was mixed with 30 μL Pseudorca crassidens plasma or Tris-buffered NaCl. PTT-A reagent (30 μL, Diagnostica Stago) was added followed by incubation for 4 minutes (37°C). Thirty microliters of 25 mM CaCl2 was added, and clotting time was measured on an ST-4 Analyzer (Diagnostica Stago).
Western blots
Plasmas (1 μL) were size-fractionated by nonreducing SDS-polyacrylamide gel electrophoresis (PAGE). Human PK, FXII, FXI, or HK were used as standards. Nitrocellulose blots were incubated with HRP-goat anti-human FXII, PK, or FXI IgG (Affinity Biologicals); goat anti-human HK IgG (Nordic Immunology); or monoclonal IgGs against FXI (14E11, 15B4)22 or FXII (1D7, 15D10).23 Monoclonal IgGs were generated by immunizing FXI-deficient or FXII-deficient Balb-C mice with mouse FXI22,24 or mouse FXII,23 and biotinylated using an EZ-link sulfo-NHS-biotinylation kit (Thermo Scientific). Anti-human HK IgG was detected with HRP-rabbit anti-goat IgG, and monoclonal IgGs with HRP-streptavidin. Blots were developed by chemiluminescence.
Protein modeling
Homology models of platypus FXI-A4 domain were generated with SWISSMODEL,25 using the human FXI crystal structure (pdb code:6i58) as a template. A dimer was generated in PyMOL Molecular Graphics System (version 1.8, Schrödinger, LLC) and checked for stereochemical quality.
Results
Background
Ancestral relationships between vertebrate groups are depicted in the cladogram in Figure 2A, with more detail regarding times of divergence provided in supplemental Figure 4. Terrestrial vertebrates (amphibians, reptiles, birds, and mammals) are called tetrapods. Tetrapods arose from lobe-finned fish ∼390 million years ago. Mammals descended from proto-mammalian synapsid reptiles, which diverged from sauropsid reptiles (ancestors of living reptiles and birds) 250 to 300 million years ago. Mammals include egg-laying (monotreme), pouched (marsupial), and placental (eutherian) forms. Example species for each vertebrate group are listed in Figure 2B and are referred to by their common names hereafter. Proteins are discussed in order of appearance in vertebrates. For this discussion, homologous proteins are orthologs (the same protein in different species) or paralogs (different proteins created by duplication of a common ancestral gene).
![Evolution of vertebrate coagulation proteins. (A) Cladogram of vertebrate evolution. Vertebrates (animals with backbones) include fish and tetrapods (land vertebrates). The earliest tetrapods were amphibians, which arose from lobe-finned fish (sarcoptyrigii) ∼390 million years ago. Amphibians are ancestral to reptiles. Mammals descended from proto-mammalian synapsid reptiles, which diverged from sauropsid reptiles (ancestors of extant reptiles and birds) ∼250 to 300 million years ago. The most primitive mammals are egg-laying monotremes, which are represented today by the duck-billed platypus and a few species of spiny anteater (echidnas). Monotremes are the ancestors of pouched (marsupial) and placental (eutherian) mammals. Cetaceans are a group of aquatic placental mammals that include whales, dolphins, and porpoises. A more detailed presentation of the estimated times of divergence for certain vertebrates is presented in supplemental Figure 4. Green lettering indicates estimated points of origin of protein components of the vitamin K-dependent thrombin generation mechanism (factors II, III [tissue factor], V, VII, VIII, IX, and X). Points of origin of the kallikrein-kinin system (FXII, PK, and HK) and FXI are indicated in blue. Red lettering and a “–” sign indicate loss of the FXII or PK genes (–FXII, –PK). Plasma factors indicated in nonitalicized lettering are plasma proteases, whereas factors indicated in italics are nonenzymatic cofactors. (B) Representative vertebrate species. The animals listed in this table are used as representatives of their respective classes in the manuscript. The abbreviations for the 5 right-hand columns are PK, FXI, FXII, HGFA (pro-HGFA), and HK. The symbols in the columns indicate if a gene for the respective proteins was identified (+) or not identified (–) in genomic analyses. There is uncertainty on whether or not a gene for Pro-HGFA is present in jawless fish (?).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/24/10.1182_bloodadvances.2020002456/1/m_advancesadv2020002456f2.png?Expires=1765912790&Signature=eiZ-Fh~PoVfbKvHbuFl4nWjqQz3Q9V1kIqaeHRsM8Z016lSZbx2fEiKLd80dms5~gc9wVAnCsBRpuyVMy4hUrlcwE~-Jlr0rxVDwY4Q5toKAYJdVRtOWXxTv-FyopGDrMDRek5XVK38JqHRYNDHjZKOWd~Fd7WTAnciI3KBY2KPyEUEuhYIUbtMMAGN6hKui~Pf4NZZifpmBTJnaQXlaQevDGq3kzMXzGD-1HoBfGlwnfwttauug36F0PuBW7AxUntT1e2CMnlvCTRyftznJHnqtGU0Fu8hNjB06~DLiSU2Po64WDZ2PkkoUmr2jH2LWxA~EuIio52op8pImBOfYlQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Evolution of vertebrate coagulation proteins. (A) Cladogram of vertebrate evolution. Vertebrates (animals with backbones) include fish and tetrapods (land vertebrates). The earliest tetrapods were amphibians, which arose from lobe-finned fish (sarcoptyrigii) ∼390 million years ago. Amphibians are ancestral to reptiles. Mammals descended from proto-mammalian synapsid reptiles, which diverged from sauropsid reptiles (ancestors of extant reptiles and birds) ∼250 to 300 million years ago. The most primitive mammals are egg-laying monotremes, which are represented today by the duck-billed platypus and a few species of spiny anteater (echidnas). Monotremes are the ancestors of pouched (marsupial) and placental (eutherian) mammals. Cetaceans are a group of aquatic placental mammals that include whales, dolphins, and porpoises. A more detailed presentation of the estimated times of divergence for certain vertebrates is presented in supplemental Figure 4. Green lettering indicates estimated points of origin of protein components of the vitamin K-dependent thrombin generation mechanism (factors II, III [tissue factor], V, VII, VIII, IX, and X). Points of origin of the kallikrein-kinin system (FXII, PK, and HK) and FXI are indicated in blue. Red lettering and a “–” sign indicate loss of the FXII or PK genes (–FXII, –PK). Plasma factors indicated in nonitalicized lettering are plasma proteases, whereas factors indicated in italics are nonenzymatic cofactors. (B) Representative vertebrate species. The animals listed in this table are used as representatives of their respective classes in the manuscript. The abbreviations for the 5 right-hand columns are PK, FXI, FXII, HGFA (pro-HGFA), and HK. The symbols in the columns indicate if a gene for the respective proteins was identified (+) or not identified (–) in genomic analyses. There is uncertainty on whether or not a gene for Pro-HGFA is present in jawless fish (?).
Evolution of vertebrate coagulation proteins. (A) Cladogram of vertebrate evolution. Vertebrates (animals with backbones) include fish and tetrapods (land vertebrates). The earliest tetrapods were amphibians, which arose from lobe-finned fish (sarcoptyrigii) ∼390 million years ago. Amphibians are ancestral to reptiles. Mammals descended from proto-mammalian synapsid reptiles, which diverged from sauropsid reptiles (ancestors of extant reptiles and birds) ∼250 to 300 million years ago. The most primitive mammals are egg-laying monotremes, which are represented today by the duck-billed platypus and a few species of spiny anteater (echidnas). Monotremes are the ancestors of pouched (marsupial) and placental (eutherian) mammals. Cetaceans are a group of aquatic placental mammals that include whales, dolphins, and porpoises. A more detailed presentation of the estimated times of divergence for certain vertebrates is presented in supplemental Figure 4. Green lettering indicates estimated points of origin of protein components of the vitamin K-dependent thrombin generation mechanism (factors II, III [tissue factor], V, VII, VIII, IX, and X). Points of origin of the kallikrein-kinin system (FXII, PK, and HK) and FXI are indicated in blue. Red lettering and a “–” sign indicate loss of the FXII or PK genes (–FXII, –PK). Plasma factors indicated in nonitalicized lettering are plasma proteases, whereas factors indicated in italics are nonenzymatic cofactors. (B) Representative vertebrate species. The animals listed in this table are used as representatives of their respective classes in the manuscript. The abbreviations for the 5 right-hand columns are PK, FXI, FXII, HGFA (pro-HGFA), and HK. The symbols in the columns indicate if a gene for the respective proteins was identified (+) or not identified (–) in genomic analyses. There is uncertainty on whether or not a gene for Pro-HGFA is present in jawless fish (?).
Origin of PK
PK is the zymogen of the protease PKa, which activates FXII and cleaves HK to liberate bradykinin.6-9 It is encoded by the Klkb1 gene. Previously, we identified Klkb1 orthologs in tetrapods, but not ray-finned fish.16 In the current analysis, Klkb1 orthologs were present in amphibian, reptile, bird, and mammal genomes, but not in cartilaginous or ray-finned fish (supplemental Figure 1). However, Klkb1 is present in 2 lobe-finned fish, the ocean-dwelling coelacanth, and the freshwater West African lungfish (Figure 2B; supplemental Figure 5). Lobe-finned fish are ancestral to tetrapods and more closely related to them in many respects than to other fish (Figure 2A).26,27 Indeed, most lungfish are obligate air breathers capable of surviving protracted periods out of water. The coelacanth represents an older lineage than lungfish that was not air breathing (supplemental Figure 4).
PK contains 4 in-tandem repeats with homology to the N-terminal domains of plasminogen and hepatocyte growth factor called apple domains (A1-A4), and a trypsin-like protease domain (Figure 3A).28-30 Kalliklectins, a group of lectins identified recently in fish, comprise 4 apple domains (supplemental Figure 6), and may be ancestral to the noncatalytic portion of PK.31,32 Each apple domain is constrained by 3 disulfide bonds (Figure 4A).29,30,33 The A4 domain of tetrapod PKs have an additional Cys321-Cys326 bond (Figure 4B; supplemental Figure 5),29 which is also present in the lungfish. PK from the more primitive coelacanth lacks Cys321-Cys326 (Figure 4B; supplemental Figure 5), similar to the A4 domain of fish kalliklectins (supplemental Figure 6). This indicates PK-A4 was originally a typical apple domain that subsequently acquired Cys321 and Cys326.
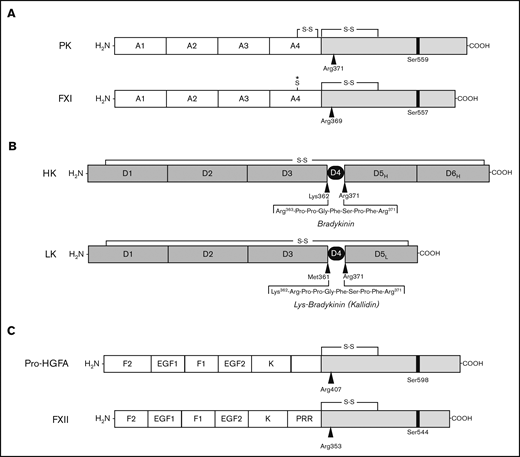
Proteins of the kallikrein-kinin system and factor XI. (A-B) Schematic diagrams of human protease precursors showing noncatalytic (white boxes) and catalytic (light gray boxes) domains. Positions of active site serine residues are indicated by black bars. Sites of proteolysis during activation are indicated by arrows. (A) Prekallikrein (PK) is a 93-kDa polypeptide that is cleaved after Arg371 to form plasma kallikrein (PKa). FXI is a homodimer of 80-kDa polypeptides. Each polypeptide is converted to FXIa by cleavage after Arg369. The noncatalytic portions of PK and FXI contain 4 apple domains, designated A1 to A4. The location of a Cys321-Cys326 intrachain disulfide bond in PK is indicated above A4. *The location of Cys321 in the A4 domain of FXI. In placental mammals and the opossum, Cys321 forms an interchain disulfide bond connecting the 2 subunits of the FXI dimer. (B) Human plasma kininogens come in high-molecular weight (HK) and low-molecular weight (LK) forms that are products of alternatively spliced messenger RNAs from the Kng1 gene. HK and LK have similar D1, D2, D3, and D4 domains, but different D5 domains (D5H and D5L, respectively). The D6 (D6H) domain is present only in HK and contains binding sites for PK and FXI. Cleavage sites for PKa in HK that release bradykinin are indicated by the black arrows. LK is cleaved at the sites indicated by black arrows by tissue kallikreins to release Lys-bradykinin (kallidin). (C) Pro-hepatocyte growth factor activator (pro-HGFA) is a 95-kDa polypeptide that is cleaved after Arg407 to form HGFA. FXII is an 80-kDa polypeptide that is converted to FXIIa by cleavage after Arg353. The Pro-HGFA and FXII noncatalytic domains are the fibronectin type 2 (F2), epidermal growth factor (EGF), fibronectin type 1 (F1), and kringle (K) domains. FXII also has a proline-rich region (PRR).
Proteins of the kallikrein-kinin system and factor XI. (A-B) Schematic diagrams of human protease precursors showing noncatalytic (white boxes) and catalytic (light gray boxes) domains. Positions of active site serine residues are indicated by black bars. Sites of proteolysis during activation are indicated by arrows. (A) Prekallikrein (PK) is a 93-kDa polypeptide that is cleaved after Arg371 to form plasma kallikrein (PKa). FXI is a homodimer of 80-kDa polypeptides. Each polypeptide is converted to FXIa by cleavage after Arg369. The noncatalytic portions of PK and FXI contain 4 apple domains, designated A1 to A4. The location of a Cys321-Cys326 intrachain disulfide bond in PK is indicated above A4. *The location of Cys321 in the A4 domain of FXI. In placental mammals and the opossum, Cys321 forms an interchain disulfide bond connecting the 2 subunits of the FXI dimer. (B) Human plasma kininogens come in high-molecular weight (HK) and low-molecular weight (LK) forms that are products of alternatively spliced messenger RNAs from the Kng1 gene. HK and LK have similar D1, D2, D3, and D4 domains, but different D5 domains (D5H and D5L, respectively). The D6 (D6H) domain is present only in HK and contains binding sites for PK and FXI. Cleavage sites for PKa in HK that release bradykinin are indicated by the black arrows. LK is cleaved at the sites indicated by black arrows by tissue kallikreins to release Lys-bradykinin (kallidin). (C) Pro-hepatocyte growth factor activator (pro-HGFA) is a 95-kDa polypeptide that is cleaved after Arg407 to form HGFA. FXII is an 80-kDa polypeptide that is converted to FXIIa by cleavage after Arg353. The Pro-HGFA and FXII noncatalytic domains are the fibronectin type 2 (F2), epidermal growth factor (EGF), fibronectin type 1 (F1), and kringle (K) domains. FXII also has a proline-rich region (PRR).
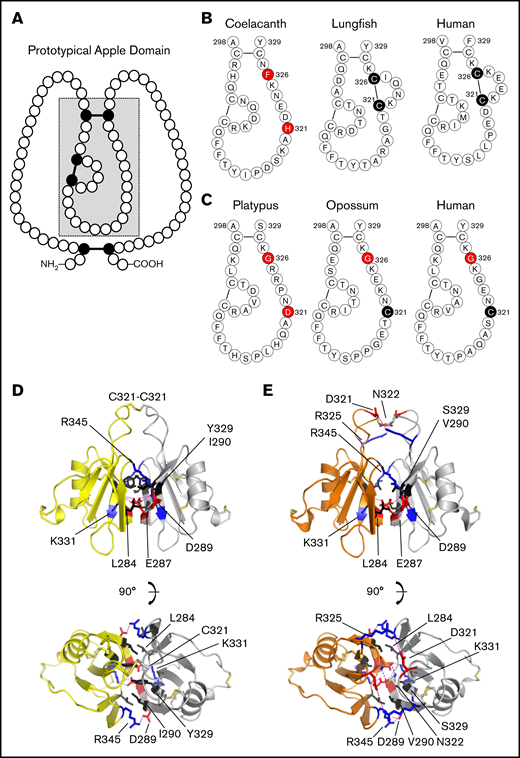
Prekallikrein and factor XI apple 4 domains. (A) Apple domain schematic. A typical apple (PAN) domain contains ∼90 amino acids (indicated by circles) and is constrained by 3 disulfide bonds (black circles and connecting bars). The region of the domain within the gray box is presented in detail in panels B and C. (B) PK-A4 residues 298 to 329. Amino acids from the PK-A4 domain highlighted by the gray box in panel A are shown for the coelacanth, West African lungfish, and human PK. Residues 321 and 326 are highlighted. In lungfish and human PK, these residues are cysteines that form a disulfide bond. In the coelacanth, they are histidine and phenylalanine. (C) FXI-A4 residues 298 to 329. Amino acids from the FXI-A4 domain highlighted by the gray box in panel A are shown for platypus, opossum, and human FXI. Residues 321 and 326 are highlighted. Residue 326 is glycine in all 3 species. In opossum and human FXI, residue 321 is a cysteine that forms the interchain disulfide bond connecting the subunits of the FXI dimer. (D) Topology diagrams of the human FXI dimer interface. Shown are 2 FXI-A4 domains (1 subunit is shown in yellow; the other in white) forming the FXI dimer interface. The Cys321-Cys321 interchain disulfide bond is shown at the top in orange. Hydrophobic residues Leu284, Ile290, and Tyr329 are shown in black, and salt bridges are formed between Lys331 (blue) and Glu287 (red) and Arg345 (blue) and Asp289 (red). The bottom image is rotated 90° relative to the top image. After Papagrigoriou et al.53 (E) Predicted FXI dimer interface for platypus FXI. Interactions are the same as those in panel D. The Cys321-Cys321 is not present in platypus FXI; instead, there is an additional salt bridge between Arg325 (blue) and Asp321 (red). The bottom image is rotated 90° relative to the top image.
Prekallikrein and factor XI apple 4 domains. (A) Apple domain schematic. A typical apple (PAN) domain contains ∼90 amino acids (indicated by circles) and is constrained by 3 disulfide bonds (black circles and connecting bars). The region of the domain within the gray box is presented in detail in panels B and C. (B) PK-A4 residues 298 to 329. Amino acids from the PK-A4 domain highlighted by the gray box in panel A are shown for the coelacanth, West African lungfish, and human PK. Residues 321 and 326 are highlighted. In lungfish and human PK, these residues are cysteines that form a disulfide bond. In the coelacanth, they are histidine and phenylalanine. (C) FXI-A4 residues 298 to 329. Amino acids from the FXI-A4 domain highlighted by the gray box in panel A are shown for platypus, opossum, and human FXI. Residues 321 and 326 are highlighted. Residue 326 is glycine in all 3 species. In opossum and human FXI, residue 321 is a cysteine that forms the interchain disulfide bond connecting the subunits of the FXI dimer. (D) Topology diagrams of the human FXI dimer interface. Shown are 2 FXI-A4 domains (1 subunit is shown in yellow; the other in white) forming the FXI dimer interface. The Cys321-Cys321 interchain disulfide bond is shown at the top in orange. Hydrophobic residues Leu284, Ile290, and Tyr329 are shown in black, and salt bridges are formed between Lys331 (blue) and Glu287 (red) and Arg345 (blue) and Asp289 (red). The bottom image is rotated 90° relative to the top image. After Papagrigoriou et al.53 (E) Predicted FXI dimer interface for platypus FXI. Interactions are the same as those in panel D. The Cys321-Cys321 is not present in platypus FXI; instead, there is an additional salt bridge between Arg325 (blue) and Asp321 (red). The bottom image is rotated 90° relative to the top image.
Origin of HK
The Kng1 gene in reptiles, birds, and mammals encodes alternatively spliced messenger RNAs for HK and low-molecular-weight kininogen (LK; Figure 3B).34,35 HK and LK share D1 through D4 domains, but have distinct D5 domains. HK also has a D6 domain that allows it to form complexes with PK and FXI in plasma.36,37 HK facilitates PK and FXI binding to surfaces, and is cleaved by PKa within D4, releasing bradykinin.6-9
Fish kininogens are more similar to mammalian LK than to HK.38-40 We did not identify D5 or fully formed D6 domains in cartilaginous or ray-finned fish. However, kininogen from the coelacanth, and probably the lungfish, has a D6 domain, and short histidine-rich motifs corresponding to D5 (supplemental Figure 7).38-40 D6 was identified in HK from all tetrapods (supplemental Figure 7), suggesting HK binds PK in most terrestrial vertebrates. HK D5 is more variable. D5 in amphibian, bird, crocodilian, and turtle HKs is shorter than in mammals and is absent in lizard and snake HKs (supplemental Figure 7).
Origin of FXII
FXII is the zymogen of the protease FXIIa, which activates PK and FXI.6-9 It is encoded by F12. FXII contains a multidomain heavy chain and a trypsin-like protease domain (Figure 3C).19,41,42 It is a paralog of pro-HGFA (encoded by HGFAC; Figure 3C; supplemental Figure 8), the precursor of the injury-response protease HGFA.16,21 HGFAC orthologs are found in all vertebrates, with some uncertainty for the most primitive jawless fish (supplemental Figure 1).1,2,16 F12 arose from a duplication of a chromosomal segment containing multiple genes, including HGFAC.16 The current analysis confirmed that F12 is present in amphibians, reptiles, and mammals, but not cartilaginous or ray-finned fish (supplemental Figure 1). Chana-Munoz et al recently identified F12 in the West African lungfish.43 We confirmed separate HGFAC and F12 genes in the lungfish (supplemental Figure 9), but did not identify F12 in the coelacanth (supplemental Figure 1). Failure to find a sequence is not proof of absence, particularly with incompletely assembled genomes. However, the results raise the possibility that PK arose before FXII.
The KKS in birds
F12 is not present in the domestic chicken,16 and studies of bird plasmas suggest FXII is missing in other avian species.44,45 Although genes flanking F12 in nonavian tetrapods are present in the chicken genome, F12 elements are not identifiable in the location occupied by F12 in nonavian tetrapods.16 This indicates F12 was inactivated, and subsequently deteriorated, in a distant ancestor of the chicken. In the current analysis, the pattern observed in the chicken was identified in all bird species evaluated (supplemental Figure 1), including primitive ratites such as ostriches and kiwis, indicating gene loss in a common avian ancestor.
In mammals, PK and FXII share an intimate relationship, undergoing reciprocal activation that is accelerated during contact activation (Figure 1B).6,42,46 Given this, it is interesting that Klkb1 is found in birds, whereas F12 is not. Previous work showed that PK is activated by FXIIa-independent mechanisms,47,48 consistent with our observation that Klkb1 may have formed before F12 in lobe-finned fish. A reasonable alternative to FXIIa as a PK activator is its paralog HGFA (Figure 3C).21 We found that HGFA does convert PK to PKa in the presence of a surface, suggesting that FXIIa may have inherited this activity from HGFA (supplemental Figure 10).
Origin of FXI
FXI is the zymogen of the protease FXIa, which activates the vitamin K-dependent coagulation protease FIX.13 FXI is encoded by F11, a gene that arose from a duplication of the gene for PK, Klkb1.16,49 FXI and PK are, therefore, paralogs (Figure 3A; supplemental Figure 11). Previously, we identified F11 in placental mammals and a marsupial (the opossum), but not in the more primitive egg-laying duck-billed platypus, and concluded that the Klkb1 duplication that created F11 occurred after marsupials diverged from monotremes.3,16 At the time, the platypus genome was not completely assembled, leading to misinterpretation on our part. In the current analysis, we identified distinct Klkb1 and F11 genes in the platypus and another monotreme, the short-beaked echidna (supplemental Figures 12 and 13). F11 is not present in amphibians, reptiles, or birds, indicating the Klkb1 duplication occurred either in a monotreme or a proto-mammalian ancestor.
Human FXI(a) and PK(a) differ in 3 key respects from a structure-function standpoint. First, FXI is a homodimer,13,50-53 whereas PK is a monomer.28-30,33 Second, FXIa is a more efficient activator of FIX than is PKa.17,18 Third, thrombin activates FXI but not PK.54-58 We compared mammalian FXI sequences to gain insights into when features unique to FXI arose.
The FXI dimer
The subunits of the human FXI dimer are connected through an interface formed by the A4 domains that is stabilized by hydrophobic interactions involving Leu284, Ile290, and Tyr329, and salt bridges between Glu287-Lys331 and Asp289-Arg345 (Figure 4D; supplemental Figure 14).13,52,53,59 Modeling indicates platypus FXI-A4 also forms these interactions and an additional Asp321-Arg325 salt bridge (Figure 4E). In placental mammals and the opossum, the Cys321-Cys321 bond covalently links the A4 domains.16,52,60 Recall that Cys321 in PK forms an intrachain bond with Cys326 (Figure 4B),29,33,60 a residue not present in FXI (Figure 4C). A simple explanation for unpaired Cys321 in FXI then would be loss of Cys326 in its PK-like ancestor. However, platypus and echidna FXI lack both Cys321 and Cys326 (Figure 4C; supplemental Figure 13). Furthermore, although Cys321 is present in the opossum (Figure 4C), a marsupial from the Western Hemisphere, Australian marsupials, like monotremes, also lack Cys321 and Cys326 (supplemental Figure 14). These data indicate that Cys321 and Cys326 were replaced early in FXI history, and that Cys321 was reintroduced after a common ancestor of the opossum and placental mammals diverged from monotremes and Australian marsupials. The Cys321-Cys321 bond is not required for FXI dimerization,52,60 and FXI Cys321 is replaced in several placental mammals, including the rabbit,61 beaver, elephant, and finless porpoise (supplemental Figure 14).
FXIa activation of FIX
Efficient FIX activation by FXIa requires a FIX-binding exosite on the FXIa-A3 domain that is not present on PKa-A3 (supplemental Figure 11).13,18,62 Although platypus FXI appears to have this exosite (supplemental Figure 14), we could not test this directly because the protein did not express in our cell culture system. Instead, we replaced A3 in human FXI with platypus FXI-A3. This protein, FXI/PlatXIA3, has identical specific activity to FXI-WT in aPTT assays (200 U/mg; supplemental Figure 15), and its active form (FXIa/PlatXIA3) activates FIX similarly to FXIa-WT (Figure 5A). In contrast, replacing FXI-A3 with human or platypus PK-A3 results in proteins (FXI/PKA3 and FXI/PlatPKA3) with activities <10% of FXI-WT (<20 U/mg; supplemental Figure 15), impaired conversion of FIX to the final product FIXaβ, and accumulation of the intermediate FIXα (Figure 5B). Thus, platypus FXI has a FIX-binding exosite, meeting an important functional criterion for FXI.

FXIa activation of FIX. (A) FIX activation by FXIa with FXI-A3 domains. Human FIX (200 nM) was incubated at 37°C with vehicle (no FXIa), 2 nM human FXIa-WT (human FXI-A3), or 2 nM human FXIa with a platypus FXI-A3 domain replacing the human A3 domain (FXIa/PlatXIA3, platypus FXI-A3). At various times, samples were removed into reducing sample buffer, size-fractionated by SDS-PAGE and stained with Coomassie blue (top row). Positions of standards for FIX, the heavy chain of the intermediate α-FIX (α), the heavy chain of the final product FIXaβ (HC), and light chain of α-FIX and FIXaβ (LC) are shown on the right of each image; positions of molecular mass markers in kilodaltons are shown to the left of the images. Stained gels underwent densitometry scanning to generate the curves in the bottom row. Values for each band were compared with those for FIX at 0 minutes, which was assigned a value of 100%. Curves show the disappearance of FIX (Δ), and the appearance of the heavy chain of the intermediate α-FIX (□), the heavy chain of FIXaβ (∇), and the light chain of α-FIX and FIXaβ (○). (B) FIX activation by FXIa with PK-A3 domains. Human FIX was incubated as in panel A with 2 nM human FXI with a human PK-A3 domain replacing the FXI A3 domain (FXIa-PKA3, human PK-A3) or human FXI with a platypus PK-A3 domain replacing the FXI A3 domain (FXIa/PlatPKA3, platypus PK-A3). Time course experiments were run and analyzed as in panel A.
FXIa activation of FIX. (A) FIX activation by FXIa with FXI-A3 domains. Human FIX (200 nM) was incubated at 37°C with vehicle (no FXIa), 2 nM human FXIa-WT (human FXI-A3), or 2 nM human FXIa with a platypus FXI-A3 domain replacing the human A3 domain (FXIa/PlatXIA3, platypus FXI-A3). At various times, samples were removed into reducing sample buffer, size-fractionated by SDS-PAGE and stained with Coomassie blue (top row). Positions of standards for FIX, the heavy chain of the intermediate α-FIX (α), the heavy chain of the final product FIXaβ (HC), and light chain of α-FIX and FIXaβ (LC) are shown on the right of each image; positions of molecular mass markers in kilodaltons are shown to the left of the images. Stained gels underwent densitometry scanning to generate the curves in the bottom row. Values for each band were compared with those for FIX at 0 minutes, which was assigned a value of 100%. Curves show the disappearance of FIX (Δ), and the appearance of the heavy chain of the intermediate α-FIX (□), the heavy chain of FIXaβ (∇), and the light chain of α-FIX and FIXaβ (○). (B) FIX activation by FXIa with PK-A3 domains. Human FIX was incubated as in panel A with 2 nM human FXI with a human PK-A3 domain replacing the FXI A3 domain (FXIa-PKA3, human PK-A3) or human FXI with a platypus PK-A3 domain replacing the FXI A3 domain (FXIa/PlatPKA3, platypus PK-A3). Time course experiments were run and analyzed as in panel A.
FXI activation
Human FXI is activated by thrombin as well as by FXIIa (Figure 6A).24,54-58 Redundant activation mechanisms may explain why FXI deficiency compromises hemostasis in humans, whereas FXII deficiency does not. The activation cleavage site in FXI from most placental mammals, including humans, has proline at the P2 position (supplemental Figure 13). This is common in thrombin substrates.58 P2 is not proline in human PK (supplemental Figure 5), which is not activated by thrombin, nor in monotreme or marsupial FXI (supplemental Figure 13). Replacing the P3 and P2 residues of human FXI with the corresponding residues from platypus FXI results in a protein (FXI-PlatRSR) activated by FXIIa but not by thrombin (Figure 6B). These data suggest that thrombin activation of FXI developed in placental mammals after they diverged from marsupials and monotremes.
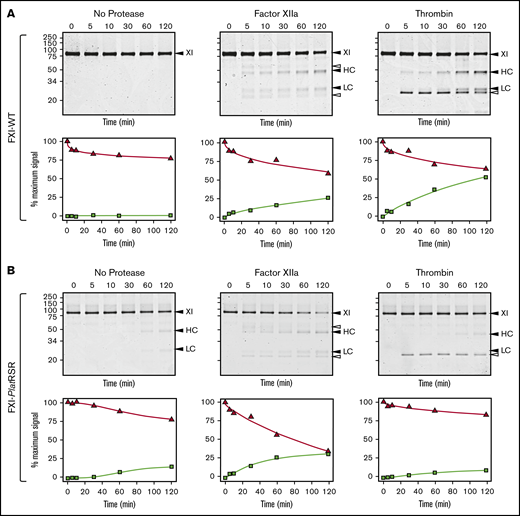
FXI activation. (A) FXI-WT activation. FXI-WT (200 nM) was incubated at 37°C with vehicle, or 40 nM FXIIa, or 140 nM thrombin in the presence of 4 μM polyphosphate. At various times, samples were removed into reducing sample buffer, size-fractionated by SDS-PAGE, and stained with Coomassie blue (top row). Positions of standards for FXI, and the heavy chain (HC) and light chain (LC) of FXIa are shown on the right of each image and positions of molecular mass markers in kilodaltons are shown to the left of the images. The white arrows indicate bands for FXIIa or thrombin that appear because of the high enzyme to substrate ratios in these reactions. Stained gels underwent densitometry scanning to generate the curves in the bottom row. Values for each band were compared with those for FXI at 0 minutes, which was assigned a value of 100%. Curves show the disappearance of FXI (Δ) and the appearance of the heavy chain of FXIa (□). (B) FXI-PlatRSR activation. FXI-PlatRSR was activated as in panel A with vehicle or with 40 nM FXIIa or 140 nM thrombin in the presence of 4 μM polyphosphate. Time course experiments were run and analyzed as in panel A.
FXI activation. (A) FXI-WT activation. FXI-WT (200 nM) was incubated at 37°C with vehicle, or 40 nM FXIIa, or 140 nM thrombin in the presence of 4 μM polyphosphate. At various times, samples were removed into reducing sample buffer, size-fractionated by SDS-PAGE, and stained with Coomassie blue (top row). Positions of standards for FXI, and the heavy chain (HC) and light chain (LC) of FXIa are shown on the right of each image and positions of molecular mass markers in kilodaltons are shown to the left of the images. The white arrows indicate bands for FXIIa or thrombin that appear because of the high enzyme to substrate ratios in these reactions. Stained gels underwent densitometry scanning to generate the curves in the bottom row. Values for each band were compared with those for FXI at 0 minutes, which was assigned a value of 100%. Curves show the disappearance of FXI (Δ) and the appearance of the heavy chain of FXIa (□). (B) FXI-PlatRSR activation. FXI-PlatRSR was activated as in panel A with vehicle or with 40 nM FXIIa or 140 nM thrombin in the presence of 4 μM polyphosphate. Time course experiments were run and analyzed as in panel A.
FXI and the KKS in cetaceans
Cetaceans are an infraorder of aquatic mammals that include whales, dolphins, and porpoises. A defect in contact activation in cetacean plasmas from loss of FXII activity was noted by Robinson and colleagues more than 50 years ago.63 Semba et al showed that the exon-intron assembly of F12 is intact in the short-finned pilot whale, but that nonsense mutations convert it to a pseudogene.64 F12 pseudogene conversion was present in all cetaceans in our study (supplemental Figure 1), consistent with published work.65-67 Huelsmann and colleagues reported recently that Klkb1 is inactive in cetaceans.67 We observed that the site adjacent to F11 normally occupied by Klkb1 in terrestrial mammals contains short sequences homologous to Klkb1 in cetacean, but no recognizable exon-intron organization. An intact Klkb1 gene is present in the hippopotamus, the terrestrial mammal most closely related to cetaceans (supplemental Figure 5), supporting a scenario in which Klkb1 and then F12 were lost in a common cetacean ancestor.
Loss of the KKS in cetaceans provides an opportunity to examine the hypothesis that FXI can function independently of FXIIa. If FXI is activated only by FXIIa, we would expect F11 to deteriorate in cetaceans because the loss of F12 would have rendered FXI nonfunctional. Some investigators reported FXI activity in cetacean plasmas,63,68 whereas others did not.69 Human plasmas lacking FXII, PK, HK, or FXI have prolonged aPTTs (Figure 7A), as does plasma from the false killer whale, a type of dolphin. In mixing studies, false killer whale plasma shortened the aPTTs of human plasmas lacking FXI or HK, but not plasmas lacking FXII or PK. Western blots of false killer whale plasma (Figure 7B) and genome analyses for other cetaceans (supplemental Figure 1) confirm the presence of HK and dimeric FXI. That these proteins persist in the absence of PK and FXII indicates they remain under selection pressure because they still perform adaptive functions.

Analysis of cetacean plasma. (A) Activated partial thromboplastin time studies. White bars show average aPTTs of pooled normal human plasma and plasma from a cetacean (a false killer whale). Gray bars show average aPTTs for plasmas from human patients lacking FXII (−XII), FXI (−XI), PK (−PK), or HK (−HK). Black bars indicate average aPTTs for mixtures of equal volumes of human factor-deficient plasmas and false killer whale plasma. Note the normal clotting times for mixtures with human plasma lacking FXI and HK. Bars represent averages of 3 runs for each plasma. (B) Plasma western blots. One-microliter samples of plasma from a false killer whale (F), a human (H), and a mouse (M) were size fractionated by SDS-PAGE and transferred to nitrocellulose. Protein controls (C) for human FXII (XII), FXI (XI), PK, or HK were included. Blots were developed with polyclonal IgGs to each human protein (left column) or monoclonal antibodies raised against mouse FXII (1D7 and 15D10, center column) or mouse FXI (14E11 and 15B4, right column). Note the absence of signals for FXII or PK, and the presence of FXI and HK, in plasma from the false killer whale.
Analysis of cetacean plasma. (A) Activated partial thromboplastin time studies. White bars show average aPTTs of pooled normal human plasma and plasma from a cetacean (a false killer whale). Gray bars show average aPTTs for plasmas from human patients lacking FXII (−XII), FXI (−XI), PK (−PK), or HK (−HK). Black bars indicate average aPTTs for mixtures of equal volumes of human factor-deficient plasmas and false killer whale plasma. Note the normal clotting times for mixtures with human plasma lacking FXI and HK. Bars represent averages of 3 runs for each plasma. (B) Plasma western blots. One-microliter samples of plasma from a false killer whale (F), a human (H), and a mouse (M) were size fractionated by SDS-PAGE and transferred to nitrocellulose. Protein controls (C) for human FXII (XII), FXI (XI), PK, or HK were included. Blots were developed with polyclonal IgGs to each human protein (left column) or monoclonal antibodies raised against mouse FXII (1D7 and 15D10, center column) or mouse FXI (14E11 and 15B4, right column). Note the absence of signals for FXII or PK, and the presence of FXI and HK, in plasma from the false killer whale.
Discussion
The original coagulation cascade represents a merger of ancient protease-based systems for thrombin and kinin generation.1,2,16 Whole and partial gene duplications drove development of both systems.1-3,16 A vitamin-K–dependent thrombin generating mechanism is found in the most primitive vertebrates (jawless fish), reflecting its vital role in hemostasis.1-3 The KKS was originally thought to have arisen in terrestrial vertebrates,16 suggesting an adaptation for life on land. However, our analysis shows that parts of the KKS (PK and HK) appeared in marine organisms (lobe-finned fish), and that a complete KKS is present in lungfish. This does not exclude the possibility that the KKS has properties that are beneficial for terrestrial life. But the system’s components appeared initially in aquatic environments. FXI, the product of a later gene duplication, connects the KKS and thrombin generation in mammals. Its absence in nonmammalian vertebrates is consistent with observations that reptile and bird plasmas clot slower than mammal plasmas in surface-initiated clotting assays.44,45,70-72 FXI is considered a coagulation factor in humans because individuals lacking it may experience excessive bleeding. However, its history as revealed by genomic analyses implies that it started as a KKS component essentially identical to PK and underwent changes that support its functions in mammalian hemostasis.
PK is a FXIIa substrate. Its active form, PKa, is a kininogenase that cleaves HK to release bradykinin.6-8 Human FXI(a) retains these activities, but has novel properties that facilitate its role in hemostasis.13,54-57 Examination of FXI sequences from extant mammals provides insight into the transition from kininogenase to coagulation protease. Our analysis indicates a FIX-binding exosite is present on the FXI-A3 domains of all living mammals, suggesting the ability to bind and efficiently activate FIX was an early feature of FXIa. Thrombin activation of FXI allows FXI to function in hemostasis independently of the KKS.54-57 Most placental mammals have proline at the FXI P2 position N-terminal to the P1 Arg369 of the activation cleavage site, an arrangement common in thrombin substrates.58 FXI from monotremes, marsupials, and a few placental mammals lack a P2 proline. Replacing the P2 residue in human FXI with the P2 serine from platypus FXI prevents activation by thrombin. These data imply that thrombin activation of FXI is a more recent acquisition than the A3 domain FIX-binding site. It seems reasonable to posit that early versions of FXI were activated similarly to the parent molecule PK. This may still be the case in primitive extant mammals such as the platypus, raising the possibility that FXIIa-mediated FXI activation is more important for hemostasis in these animals than in placental mammals. The impression that the KKS does not play a role in hemostasis in placental mammals therefore may reflect the acquisition of an alternative mechanism for FXI activation that places the protein partly under control of the thrombin generation mechanism. This transition has been taken furthest in cetaceans, in which FXI must function without the KKS because of the loss of FXII and PK.
It could be argued that the KKS likely had no role in thrombin generation before the arrival of FXI. However, Osterud and colleagues showed more than 40 years ago that PKa activates FIX.73,74 The reaction is inefficient by FXIa standards (100-fold lower catalytic efficiency kcat/Km)17,18,62 ; however, Visser et al recently showed that PKa contributes to FIX activation independently of FXI during contact activation in plasma.75 Noubouossie et al reported erythrocyte microvesicles support PKa-mediated FIX activation.76 These observations raise the possibility that PKa in nonmammalian tetrapods contributes to thrombin generation through FIX activation. The development of FXI in mammals may therefore represent enhancement of an activity inherent in the KKS, rather than de novo formation of a FIX activation mechanism. Bleeding in FXI-deficient humans is highly variable.14 Perhaps the effects of PKa-mediated FIX activation, normally masked by FXI, can compensate for absence of FXI in some individuals.
The impression that the KKS is not required for hemostasis in mammals is based on observations of humans and domesticated animals during surgery or after trauma, and experience with animals in the laboratory. Juang and colleagues recently suggested FXII contributes to mammalian hemostasis outside these settings.77 They noted that bleeding duration and blood loss after injury are significantly reduced in WT mice, but not FXII-deficient mice, if wounds are exposed to silicate-containing soils. Silicates are major constituents of the Earth’s crust, and are used to trigger clotting in aPTT assays. Most tetrapods spend their lives in close contact with silicate-rich soil, and soil contamination of wounds would be common. This applies to lungfish, the most primitive organism in which FXII has been identified. Hemostasis following injury that does not involve soil contamination, on the other hand, would not appear to require FXII because a key cofactor for its activation, silicate, is missing. It is interesting in this regard that FXII has been lost in birds and cetaceans, vertebrates that probably spend less time in contact with soil than other tetrapods.
Reciprocal conversion of PK and FXII to PKa and FXIIa appears to contribute to basal bradykinin formation in mammals.46,78 Revenko and coworkers noted that reducing the plasma level of either PK or FXII in healthy mice leads to decreased baseline activation of the other protein.46 However, there is evidence for PK activation independent of FXII. Shariat-Madar et al showed that human PK is activated by the lysosomal protease prolyl-carboxypeptidase.47 Joseph et al showed that the chaperone HSP90 promotes PK conversion to PKa.48 The absence of FXII in birds, the possible appearance of PK before FXII in lobe-finned fish, and our demonstration that PK is activated by the FXIIa paralog HGFA, support the premise that PK can function independently of FXIIa. These findings illustrate an important point regarding the unimpressive phenotypes of mammals, including humans, deficient in individual components of the KKS. The apparent absence of defects in hemostasis or other processes may be due to the presence of redundant or overlapping systems. Just as alternative mechanisms for FIX activation obviate the requirement for FXI in some individuals, and thrombin activation of FXI may mask the contribution of the KKS to clotting, multiple processes for PK activation may reduce reliance on FXIIa as a PK activator.
Recombinant proteins described in the manuscript will be made available to other investigators upon request to the corresponding author, David Gailani, at e-mail: dave.gailani@vanderbilt.edu. All genomic data analyzed are available on public Web sites; however, the authors would be happy to supply interested parties with the downloaded data used in this study upon request.
Acknowledgments
This manuscript is dedicated to the memory of our colleague and friend Russell F. Doolittle, whose pioneering studies on the evolution of blood coagulation are the inspiration for the work presented here.
This study was supported by a Dean’s Faculty of Biology and Environmental Protection travel subsidy and grant (no. 506/1136) from the University of Lodz, Poland (M.B.P.); Program Grant (RG/12/9/29775) from the British Heart Foundation (J.E.); and a grant from the National Institutes of Health, National Heart, Lung, and Blood Institute (HL140025) (D.G.).
Authorship
Contribution: M.B.P. conducted genomic, structural, and evolutionary analyses and contributed to writing the manuscript; A.S. and M.-f.S. prepared and studied recombinant FXII and Pro-HGFA, and studied coagulation in cetacean plasma; A.L., S.K.D., and P.S. designed and prepared recombinant FXI and FXI chimeras and tested them in protease and coagulation assays; C.K. designed experiments with cetacean plasma and contributed to writing the manuscript; A.G. characterized monoclonal antibodies to mammalian FXI and FXII and contributed to writing the manuscript; J.E. conducted modeling of FXI dimer, and contributed to genomic analyses and writing the manuscript; and D.G. oversaw the project and writing of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: David Gailani, Vanderbilt University Medical Center, Room 4918, The Vanderbilt Clinic, 1301 Medical Center Dr, Nashville, TN 37232; e-mail: dave.gailani@vanderbilt.edu.

