


Abstract
T cells engineered with chimeric antigen receptors (CARs) have revolutionized the field of cell therapy and changed the paradigm of treatment for many patients with relapsed or refractory B-cell malignancies. Despite this progress, there are limitations to CAR-T cell therapy in both the autologous and allogeneic settings, including practical, logistical, and toxicity issues. Given these concerns, there is a rapidly growing interest in natural killer cells as alternative vehicles for CAR engineering, given their unique biological features and their established safety profile in the allogeneic setting. Other immune effector cells, such as invariant natural killer T cells, γδ T cells, and macrophages, are attracting interest as well and eventually may be added to the repertoire of engineered cell therapies against cancer. The pace of these developments will undoubtedly benefit from multiple innovative technologies, such as the CRISPR-Cas gene editing system, which offers great potential to enhance the natural ability of immune effector cells to eliminate refractory cancers.
Clinical case
A 46-year-old woman with no previous medical problems presented to her primary care physician with complaints of neck swelling and pressure in her throat. She denied any history of fever, night sweats, or weight loss. On physical examination she was noted to have palpable lymph nodes in the neck and inguinal areas. Computed tomography scanning of the neck, chest, abdomen, and pelvis showed diffuse lymphadenopathy above and below the diaphragm. Laboratory values revealed a hemoglobin of 11 g/dL and a lactate dehydrogenase of 431 U/L. Excisional biopsy of a left inguinal lymph node and a bone marrow biopsy confirmed the diagnosis of grade 3, stage IV follicular lymphoma with bone marrow involvement. The Follicular Lymphoma International Prognostic Index score was 4, indicating high-risk disease. After receiving 6 cycles of rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone chemotherapy, the patient achieved a complete remission. Four years later, she relapsed and was treated with multiple lines of therapy, including rituximab, obinutuzumab plus bendamustine, and rituximab, gemcitabine, and oxaliplatin. The treatments were ineffective, and the disease became refractory, with the patient entering a leukemic phase with leukocytosis (white blood cells >200 × 103/μL with 90% lymphocytes). A positron emission tomography–computed tomography scan showed increased fluorodeoxyglucose uptake (up to a standardized uptake value of 14) in multiple lymph nodes above and below the diaphragm, with bulky abdominal lymphadenopathy. Biopsy of an inguinal lymph node showed follicular lymphoma grade 2 (90%) and grade 3A (10%). Bone marrow biopsy revealed extensive involvement with follicular lymphoma, and flow cytometry showed an aberrant λ-restricted B-cell population positive for CD19, CD20, CD22, CD38 dim, and CD10 dim and negative for CD5, CD43, and CD200. The patient was treated with hyperfractionated cyclophosphamide plus dexamethasone and achieved a partial response, although persistent bulky abdominal lymph nodes were still apparent.
CAR-T cell therapy: advantages and limitations
T cells modified to express a chimeric antigen receptor (CAR) represent a major advance in the fields of cell therapy and personalized medicine.1 In this strategy, a patient’s own T cells are isolated and engineered to express a synthetic receptor that binds a tumor antigen to induce tumor cell death. These CAR-engineered T cells are then expanded ex vivo to clinically significant numbers and infused back into the patient as cancer immunotherapy. The potency of these engineered cells lies in merging the effector functions of T lymphocytes with the specificity and binding affinity of antibodies. The extracellular domain of a CAR comprises an antigen‐binding single-chain variable fragment made up of the variable heavy and variable light chains of an antibody, fused by a short peptide linker.2 The intracellular domain consists of a signaling molecule, traditionally from the T-cell receptor (TCR) CD3ζ chain, and other (optional) features depending on the generation of the CAR construct.2 Whereas first-generation CARs contain CD3ζ alone, second-generation CARs incorporate an additional costimulatory endodomain, such as CD28 or 4‐1BB, and third-generation CARs contain >1 costimulatory domain fused to CD3ζ.1 Finally, fourth-generation CARs harbor an extra transgenic payload such as cytokines to boost their effector function.3-5
CAR-T cells were first tried against B-cell malignancies with CD19 used as a target antigen, resulting in remarkable clinical responses in diseases that were multiply relapsed and refractory to chemotherapy.6 This success led to the US Food and Drug Administration approval of 2 autologous CAR-T cell products: tisagenlecleucel (Kymriah) and axicabtagene ciloleucel (Yescarta).7-9 Kymriah was approved for patients ≤25 years of age with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL) based on the results from the phase 2 pivotal ELIANA trial that reported an overall response rate (ORR) of 81%, with 60% of patients having achieved complete remission (CR).7 Kymriah is also approved for adults with relapsed or refractory large B-cell lymphoma, including patients with diffuse large B-cell lymphoma (DLBCL) not otherwise specified, high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma for whom ≥2 lines of systemic therapy have failed, based on the JULIET trial reporting an ORR of 52% with a CR rate of 40%.9 The ZUMA-1 trial led to the approval of Yescarta for use for adult patients with large B-cell lymphoma after ≥2 lines of therapy, including DLBCL, primary mediastinal large B-cell lymphoma, high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma. The trial reported an ORR of 82% and a CR rate of 54%, with ongoing responses observed in 42% of patients.8
Despite the elegance of this therapy and its clinical successes, autologous CAR-T cells have some limitations.10 First and foremost, from a clinical standpoint, not all patients can be candidates for this therapy. For example, some patients with cancer are heavily pretreated and have significant T-cell lymphopenia, which could potentially hamper collection of autologous T cells in sufficient numbers for a clinically relevant dose of CAR-T cells.11 Moreover, the process of producing an autologous CAR-T cell product is lengthy and logistically cumbersome; therefore, patients who have rapidly progressive disease are usually not candidates for this therapy. Another limitation of CAR-T cells is their toxicity profile, because clinical experience has shown a substantial risk of cytokine release syndrome (CRS) and immune effector cell–associated neurotoxicity syndrome (ICANS) among patients receiving this therapy,12 especially those with bulky or high-burden disease.13,14 Fortunately, advances in consensus grading and management guidelines for these inflammatory syndromes have significantly improved outcomes for patients with CAR-T cell–related toxicities.15 Target antigen loss after CAR-T cell therapy can pose another clinical problem, because CAR-T cell killing depends on CAR engagement; that is, target antigen loss renders these immune cells ineffective.16 Finally, allogeneic CAR-T cells are being explored for off-the-shelf therapy,17,18 but given their associated risk of graft-versus-host disease (GVHD), allogeneic CAR-T cells would need further genetic modification, such as deletion of the TCR, to mitigate this risk.19
Back to our patient
Given the advanced stage of her disease, which had resisted multiple lines of therapy, and the leukemic phase of her follicular lymphoma, which predicts poor prognosis,20 our patient was considered for treatment with autologous CAR-T cell therapy. However, she was not a candidate for this therapy because Yescarta and Kymriah are approved only for follicular lymphoma that has transformed to high-grade lymphoma, and even if the disease did fit the transformation criteria, the patient’s severe T-cell lymphopenia and rapidly progressing disease would not permit the collection of autologous T cells and the necessary wait for the manufacture of an autologous CAR-T cell product. Next, the patient was considered for a clinical trial of allogeneic CAR-T cell therapy but was not eligible in view of multiple antibodies against the mismatched HLA alleles with the donors. In addition, our patient had bulky abdominal disease, which would have led to an increased risk of CRS and ICANS with use of CAR-T cell therapy.21
NK cells as an alternative platform for CAR engineering
In view of the aforementioned limitations of CAR-T cell therapy and of the patient’s specific clinical and disease-related features, a decision was made to take an alternative approach using CAR natural killer (NK) cells that might circumvent these problems.
NK cells are CD3-CD56+ innate lymphoid cells that play a fundamental role in host defenses against infectious agents and malignancies.22 Unlike T cells, NK cells can kill transformed cells without the need for prior antigen priming, and their killing capacity is not restricted by the target cell’s expression of major histocompatibility complex (MHC) molecules.23 In fact, NK cell activity is governed by a broad repertoire of activating and inhibitory receptors (Figure 1) whose complex integration of positive and negative signals determines the final disposition of NK cells to kill or not to kill a target cell.24 As a result, NK cells are capable of distinguishing between normal and “stressed” cells. Healthy cells are spared as they express self-MHC class I molecules that bind to inhibitory receptors on NK cells, thus delivering a “no kill” signal, whereas transformed or infected cells that downregulate MHC class I molecules or upregulate stress-induced molecules such as MICA/MICB and ULBP, which bind to activating receptors such as NKG2D, deliver an activating signal to the NK cells to kill.25 Thus, tumor cells that escape T-cell killing by downregulating MHC class I are still susceptible to NK cell killing. Another major advantage of NK cells over T cells is that they do not cause GVHD in the allogeneic setting,26 making them a safe choice as a third-party, off-the-shelf cellular therapy option. Allogeneic NK cells may also be less prone to rejection by recipient alloreactive T cells. A number of preclinical studies have shown that NK cells in the graft can target and kill host lymphohematopoietic cells that may participate in the rejection of donor cells.27 Indeed, our clinical data confirm the persistence of adoptively transferred CAR-NK cells at low levels in patients for at least a year, despite HLA mismatching,26 supporting the notion that NK cells may be less susceptible to host-versus-graft rejection. Therefore, these favorable features of NK cells with regard to GVHD and host-versus-graft reactions may obviate HLA matching in the allogeneic setting.
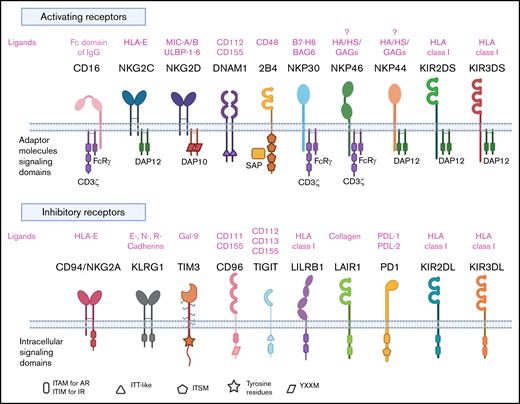
NK cell repertoire of activating and inhibitory receptors. AR, activating receptor; BAG6, BCL2-associated athanogene 6; DAP10, DNAX activating protein of 12 KDa; DAP12, DNAX activating protein of 12 KDa; DNAM1, DNAX accessory molecule 1; GAGs, glycosaminoglycans; Gal-9, galectin-9; HA, hemagglutinin; HS, heparan sulfate; IR, inhibitory receptor; ITAM, immunoreceptor tyrosine-based activation motif; ITIM, immunoreceptor tyrosine-based activation motif; ITSM, immunoreceptor tyrosine-based switch motif; ITT-like, immunoglobulin tail tyrosine-like; KIR, killer immunoglobulin like receptor; KLRG1, killer cell lectin-like receptor G1; LAIR1, leukocyte-associated immunoglobulin like receptor-1; LILRB1, leukocyte Ig-like receptor B1; MIC-A/B, MHC class I chain-related proteins A and B; PD1, programmed cell death protein 1; PDL-1, programmed cell death ligand 1; PDL-2, programmed cell death ligand 2; TIGIT, T-cell immunoreceptor with Ig and ITIM domains; TIM3, T-cell immunoglobulin mucin domain-3; YXXM, Y stands for tyrosine, X for any amino acid residue, and M for methionine.
NK cell repertoire of activating and inhibitory receptors. AR, activating receptor; BAG6, BCL2-associated athanogene 6; DAP10, DNAX activating protein of 12 KDa; DAP12, DNAX activating protein of 12 KDa; DNAM1, DNAX accessory molecule 1; GAGs, glycosaminoglycans; Gal-9, galectin-9; HA, hemagglutinin; HS, heparan sulfate; IR, inhibitory receptor; ITAM, immunoreceptor tyrosine-based activation motif; ITIM, immunoreceptor tyrosine-based activation motif; ITSM, immunoreceptor tyrosine-based switch motif; ITT-like, immunoglobulin tail tyrosine-like; KIR, killer immunoglobulin like receptor; KLRG1, killer cell lectin-like receptor G1; LAIR1, leukocyte-associated immunoglobulin like receptor-1; LILRB1, leukocyte Ig-like receptor B1; MIC-A/B, MHC class I chain-related proteins A and B; PD1, programmed cell death protein 1; PDL-1, programmed cell death ligand 1; PDL-2, programmed cell death ligand 2; TIGIT, T-cell immunoreceptor with Ig and ITIM domains; TIM3, T-cell immunoglobulin mucin domain-3; YXXM, Y stands for tyrosine, X for any amino acid residue, and M for methionine.
Despite their advantages, NK cells have a number of limitations that could affect their efficacy. These include a short lifespan of only 1 to 2 weeks in the absence of cytokine support, limited cell numbers often requiring ex vivo expansion and activation, and, in common with other immune cells, susceptibility to the immunosuppressive tumor microenvironment that could in turn limit their trafficking and effector function. Advances in engineering have enabled investigators to overcome some of these limitations. For example, the incorporation of cytokine transgenes (eg, interleukin [IL]-2 or IL-15) in NK cells can enhance their proliferation and persistence, and the incorporation of chemokine receptors can promote their trafficking to tumor sites.25 Other engineering strategies to improve NK cell performance are reviewed elsewhere.25,28
In view of their unique biological features, their potent innate antitumor activity, and their favorable safety profile in the clinic,25,26 NK cells have garnered considerable attention over the past few years as an emerging alternative platform for CAR engineering. Pure populations of NK cells can be derived from autologous or allogeneic sources, such as peripheral blood; umbilical cord blood; stem cells, including induced pluripotent stem cells and hematopoietic stem cells; and in vitro propagated NK cell lines such as NK-92.29 Limitations of autologous NK cells derived from patients with cancer, including reduced effector function and the need for a patient-specific product (similar to autologous T cells), have led to the rise of allogeneic NK cells as a platform for CAR engineering.30 Allogeneic NK cells used for CAR engineering can be derived from multiple sources, each with unique advantages and limitations.29 Cord blood is a readily available off-the-shelf source of allogeneic NK cells, which, though numerically few, can be expanded to large, highly functional doses because of their inherently high proliferative capacity.31 In addition, access to hundreds of thousands of cord blood units in the global inventories allows selection of units without cross-reactivity to allosensitized patients, as for the case presented here. Peripheral blood NK cells, on the other hand, are phenotypically mature and highly functional; however, their use requires a willing healthy donor to undergo leukapheresis or blood donation. NK cells derived from induced pluripotent stem cells are immature, with low expression of antibody-dependent cellular cytotoxicity–inducing CD16 receptors, but are highly proliferative and can be made readily available for use in biobanks.32 Finally, NK cell lines such as NK-92 cells can be easily expanded and manipulated, but because they originate from undifferentiated NK cell precursors from patients with NK lymphoma, they lack expression of CD16 and some killer immunoglobulin-like receptors, and because of their malignant potential they need irradiation before clinical use, which in turn could limit their in vivo persistence and efficacy.33 A multitude of preclinical studies have confirmed the activity of CAR-engineered NK cells derived from these sources against a range of cancer models, both in vitro and in vivo (reviewed by Pfefferle et al).29 More recently, the efficacy of CAR-NK cells is being explored in different malignancies (completed or recruiting clinical studies are summarized in Table 1). We conducted the first-in-human phase 1/2 clinical trial of CAR-NK cell therapy for patients with relapsed or refractory B-cell hematologic malignancies (ClinicalTrials.gov number NCT03056339).26 The NK cells were derived from cord blood and were HLA mismatched with the recipient. The retroviral vector used to transduce the NK cells encoded a CAR against the CD19 antigen and IL-15 to enhance NK cell persistence and function. An inducible caspase 9 suicide gene (iCas9) was added as a safety switch.26 Eleven heavily pretreated patients have received this therapy at 3 different dose levels (1 × 105, 1 × 106, or 1 × 107 cells per kilogram). Eight responded, for an overall response rate of 73%, with 7 achieving a complete remission (64%). These responses were rapid and seen at all dosage levels. Importantly, serious toxicities including CRS, ICANS, or GVHD did not develop in any of the patients.26 Based on these data, our patient is being considered for participation in the CAR-NK cell study. A schematic diagram of the ultimate goal of producing off-the-shelf cord blood derived CAR-NK cells for the treatment of patients with cancer is represented in panel A of the visual abstract.
Clinical trials evaluating alternative immune cells for CAR-based cancer therapy
| National Clinical Trial identifier | Clinical trial phase | Cancer type | Antigen target | NK cell source | Construct/method | Dosage | Status | Location |
|---|---|---|---|---|---|---|---|---|
| Active CAR NK cell trials | ||||||||
| NCT03056339 | 1/2 | B-lymphoid malignancies, ALL, CLL, NHL | CD19 | Cord blood | CAR.CD19-CD28-CD3ζ-iCasp9-IL15 | 3 dosage levels: | Phase 1 portion completed, phase 2 recruiting | MD Anderson Cancer Center, Houston, TX USA |
| 105/kg | ||||||||
| 106/kg | ||||||||
| 107/kg | ||||||||
| NCT00995137 | 1 | B-ALL | CD19 | Haploidentical donor | CAR.19-41BB-CD3ζ | Unknown | Completed | St Jude Children’s Research Hospital, Memphis, TN |
| NCT04245722 | 1 | B-cell lymphoma, CLL | CD19 ± CD20 antibody (rituximab or obinutuzumab) | Induced pluripotent stem cell–derived NK cells | CAR.19-NKG2D-2B4-CD3ζ-IL15RF-hnCD16 | Dose escalation, exact dosages unknown | Recruiting | University of Minnesota Masonic Cancer Center, Minneapolis, MN |
| NCT03940833 | 1/2 | Multiple myeloma | BCMA | NK-92 cell line | Unknown | Unknown | Recruiting | China |
| NCT03415100 | 1 | Solid tumors | NKG2D ligands | Autologous or allogeneic NK | mRNA electroporation | Unknown | Recruiting | China |
| NCT03940820 | 1/2 | Solid tumors | ROBO1 | Unknown | Unknown | Recruiting | China | |
| NCT03941457 | 1/2 | Pancreatic cancer | ROBO1 | Unknown | Unknown | Unknown | Recruiting | China |
| NCT03383978 | 1 | Glioblastoma | HER2 | NK-92 | CAR.HER2-CD28- CD3ζ | 1 × 107-1 × 108 intracranial infusion | Recruiting | Germany |
| CAR-NK T cell trials | ||||||||
| NCT03774654 | 1 | Relapsed or refractory B-cell malignancies | CD19 | Allogeneic iNKT cells | CAR.19-CD28- CD3ζ -IL15 | 4 dosage levels: | Not yet recruiting | Baylor Methodist–Texas Children’s, Houston, TX |
| 1 × 107/m2 | ||||||||
| 3 × 107/m2 | ||||||||
| 1 × 108/m2 | ||||||||
| 3 × 108/m2 | ||||||||
| NCT03294954 | 1 | Relapsed or refractory neuroblastoma | GD2 | Autologous iNKT cells | CAR.GD2-CD28-CD3ζ-IL15 | 4 dosage levels: | Recruiting | Baylor Methodist–Texas Children’s, Houston, TX |
| 3 × 106/m2 | ||||||||
| 1 × 107/m2 | ||||||||
| 3 × 107/m2 | ||||||||
| 1 × 108/m2 | ||||||||
| CAR γδ T cell trials | ||||||||
| NCT02656147 | 1 | B-cell leukemia and lymphoma | CD19 | Allogeneic | Unknown | Unknown | Not yet recruiting | China |
| γδ T cells | ||||||||
| NCT04107142 | 1 | Solid tumors | NKG2D ligands | Haploidentical or allogeneic γδ T cells | Unknown | 3 × 108- | Not yet recruiting | Malaysia |
| 3 × 109 cells | ||||||||
| CAR CIK trial | ||||||||
| NCT03389035 | 1/2 | Relapsed B-ALL | CD19 | Allogeneic (donor derived peripheral blood) CIK | CAR.19-CD28-OX40- CD3ζ | 4 dose levels: 1 × 106, 3 × 106 | Recruiting | Italy |
| Sleeping beauty transposon | 7.5 × 106 | |||||||
| 15 × 106 | ||||||||
| CAR+ cells/kg | ||||||||
| National Clinical Trial identifier | Clinical trial phase | Cancer type | Antigen target | NK cell source | Construct/method | Dosage | Status | Location |
|---|---|---|---|---|---|---|---|---|
| Active CAR NK cell trials | ||||||||
| NCT03056339 | 1/2 | B-lymphoid malignancies, ALL, CLL, NHL | CD19 | Cord blood | CAR.CD19-CD28-CD3ζ-iCasp9-IL15 | 3 dosage levels: | Phase 1 portion completed, phase 2 recruiting | MD Anderson Cancer Center, Houston, TX USA |
| 105/kg | ||||||||
| 106/kg | ||||||||
| 107/kg | ||||||||
| NCT00995137 | 1 | B-ALL | CD19 | Haploidentical donor | CAR.19-41BB-CD3ζ | Unknown | Completed | St Jude Children’s Research Hospital, Memphis, TN |
| NCT04245722 | 1 | B-cell lymphoma, CLL | CD19 ± CD20 antibody (rituximab or obinutuzumab) | Induced pluripotent stem cell–derived NK cells | CAR.19-NKG2D-2B4-CD3ζ-IL15RF-hnCD16 | Dose escalation, exact dosages unknown | Recruiting | University of Minnesota Masonic Cancer Center, Minneapolis, MN |
| NCT03940833 | 1/2 | Multiple myeloma | BCMA | NK-92 cell line | Unknown | Unknown | Recruiting | China |
| NCT03415100 | 1 | Solid tumors | NKG2D ligands | Autologous or allogeneic NK | mRNA electroporation | Unknown | Recruiting | China |
| NCT03940820 | 1/2 | Solid tumors | ROBO1 | Unknown | Unknown | Recruiting | China | |
| NCT03941457 | 1/2 | Pancreatic cancer | ROBO1 | Unknown | Unknown | Unknown | Recruiting | China |
| NCT03383978 | 1 | Glioblastoma | HER2 | NK-92 | CAR.HER2-CD28- CD3ζ | 1 × 107-1 × 108 intracranial infusion | Recruiting | Germany |
| CAR-NK T cell trials | ||||||||
| NCT03774654 | 1 | Relapsed or refractory B-cell malignancies | CD19 | Allogeneic iNKT cells | CAR.19-CD28- CD3ζ -IL15 | 4 dosage levels: | Not yet recruiting | Baylor Methodist–Texas Children’s, Houston, TX |
| 1 × 107/m2 | ||||||||
| 3 × 107/m2 | ||||||||
| 1 × 108/m2 | ||||||||
| 3 × 108/m2 | ||||||||
| NCT03294954 | 1 | Relapsed or refractory neuroblastoma | GD2 | Autologous iNKT cells | CAR.GD2-CD28-CD3ζ-IL15 | 4 dosage levels: | Recruiting | Baylor Methodist–Texas Children’s, Houston, TX |
| 3 × 106/m2 | ||||||||
| 1 × 107/m2 | ||||||||
| 3 × 107/m2 | ||||||||
| 1 × 108/m2 | ||||||||
| CAR γδ T cell trials | ||||||||
| NCT02656147 | 1 | B-cell leukemia and lymphoma | CD19 | Allogeneic | Unknown | Unknown | Not yet recruiting | China |
| γδ T cells | ||||||||
| NCT04107142 | 1 | Solid tumors | NKG2D ligands | Haploidentical or allogeneic γδ T cells | Unknown | 3 × 108- | Not yet recruiting | Malaysia |
| 3 × 109 cells | ||||||||
| CAR CIK trial | ||||||||
| NCT03389035 | 1/2 | Relapsed B-ALL | CD19 | Allogeneic (donor derived peripheral blood) CIK | CAR.19-CD28-OX40- CD3ζ | 4 dose levels: 1 × 106, 3 × 106 | Recruiting | Italy |
| Sleeping beauty transposon | 7.5 × 106 | |||||||
| 15 × 106 | ||||||||
| CAR+ cells/kg | ||||||||
ALL, acute lymphoblastic leukemia; BCMA, B-cell maturation antigen; CLL, chronic lymphocytic leukemia; iCasp9, inducible caspase 9; mRNA, messenger RNA; NHL, non-Hodgkin lymphoma.
Other immune effector cells as vehicles for CAR engineering
Immune effectors other than NK cells are also being investigated as alternative platforms for CAR engineering. These cell populations possess a number of specific biological features that could significantly expand and diversify the repertoire of CAR-based therapies. For example, invariant NK T cells have attracted growing attention as possible CAR vehicles, because they possess features of both innate and adaptive immune cells. Much like innate immune cells, they can mount a rapid response to antigen exposure, but they can also display precise antigen recognition in the manner of adaptive cells.34 Unlike conventional T cells, their TCR recognizes lipid antigens presented by CD1d, a monomorphic MHC class 1–like molecule.34 Another group of immune effector cells being explored as potential platforms for CAR engineering are γδ T cells. These T cells are predominant at epithelial surfaces and express γδ TCRs, which are triggered in an MHC-independent fashion (eg, by aminobisphosphonates), contrary to αβ TCR activation.35 γδ T cells can also cross-present antigens to αβ T cells and thus can serve as a link between innate and adaptive immunity.36 Cytokine-induced killer cells (CIK) are a group of immune effector cells featuring a mixed T and NK cell–like phenotype and therefore can kill tumor targets in both MHC-dependent and MHC-independent manners.37,38 Preclinical studies exploring CAR-transduced CIK cells have reported promising results both in hematological39,40 and solid tumor41-43 settings and have led to an ongoing clinical trial (NCT03389035) to test the safety of CAR CIK-CD19 cells in adult and pediatric patients with relapsed or refractory B-cell acute lymphoblastic leukemia. Other than lymphocytes, a different type of immune cell that offers the advantage of being able to penetrate tumor beds and naturally engulf malignant cells, macrophages, has garnered attention as a possible effector in CAR-based therapies.44 The advantages and disadvantages of alternative immune effector cells for CAR-based cancer therapy are summarized in Table 2. Preclinical evaluations of these types of immune cells as CAR platforms support this prediction (reviewed by Rotolo et al45 ), and clinical trials using these cell types are either planned or ongoing (see Table 1).
Advantages and disadvantages of alternative immune effector cells as platforms for CAR engineering
| Alternative immune effector cell | Advantages | Disadvantages | Safety profile |
|---|---|---|---|
| NK cell | Multiple innate activating receptors that can mediate killing | Low persistence in the absence of cytokine | In early clinical results of CAR-NK cells: |
| Can harness KIR-ligand mismatch and “missing self” to reduce risk of relapse | Numerically few necessitating ex vivo expansion | -No GVHD | |
| Multiple mechanisms of cytotoxicity | Suboptimal trafficking and penetration into solid tumors | -No CRS | |
| No need for previous antigen priming | -No ICANS | ||
| Rapid tumor killing | |||
| iNKT | Innate and adaptive features | Can have immunosuppressive properties (Th2, Th17) | Limited clinical data with iNKT-CAR NK cells; reports in 2 patients showed no toxicity |
| Invariant TCR recognizes lipid antigens presented by CD1d | Numerically few requiring ex vivo expansion | In non–CAR-engineered cells: | |
| -No GVHD | |||
| -No toxicities | |||
| γδ T cells | Links innate and adaptive immune systems | Can have immunosuppressive properties (γδ T17, Vδ1 γδ T cells, γδ Treg) | No clinical data with CAR γδ T cells |
| MHC independent γδ TCR | Numerically few necessitating ex vivo expansion | In non–CAR-engineered cells: | |
| Cross-present antigens to αβ T cells | No GVHD | ||
| No toxicities | |||
| Macrophages | Good penetration into solid tumors | Can have immunosuppressive properties (M2) | No clinical data with CAR macrophages |
| Mediates phagocytosis and cytotoxicity | Numerically few necessitating ex vivo expansion | In non–CAR-engineered cells: | |
| Cross present antigens to αβ T cells | No GVHD | ||
| No toxicities | |||
| CIK | Multiple killing mechanisms including MHC-dependent and MHC-independent | Heterogeneous products | No clinical data with CAR CIK |
| Numerically few necessitating ex vivo expansion | In non–CAR-engineered cells: | ||
| Lower GVHD risk than T cells60 |
| Alternative immune effector cell | Advantages | Disadvantages | Safety profile |
|---|---|---|---|
| NK cell | Multiple innate activating receptors that can mediate killing | Low persistence in the absence of cytokine | In early clinical results of CAR-NK cells: |
| Can harness KIR-ligand mismatch and “missing self” to reduce risk of relapse | Numerically few necessitating ex vivo expansion | -No GVHD | |
| Multiple mechanisms of cytotoxicity | Suboptimal trafficking and penetration into solid tumors | -No CRS | |
| No need for previous antigen priming | -No ICANS | ||
| Rapid tumor killing | |||
| iNKT | Innate and adaptive features | Can have immunosuppressive properties (Th2, Th17) | Limited clinical data with iNKT-CAR NK cells; reports in 2 patients showed no toxicity |
| Invariant TCR recognizes lipid antigens presented by CD1d | Numerically few requiring ex vivo expansion | In non–CAR-engineered cells: | |
| -No GVHD | |||
| -No toxicities | |||
| γδ T cells | Links innate and adaptive immune systems | Can have immunosuppressive properties (γδ T17, Vδ1 γδ T cells, γδ Treg) | No clinical data with CAR γδ T cells |
| MHC independent γδ TCR | Numerically few necessitating ex vivo expansion | In non–CAR-engineered cells: | |
| Cross-present antigens to αβ T cells | No GVHD | ||
| No toxicities | |||
| Macrophages | Good penetration into solid tumors | Can have immunosuppressive properties (M2) | No clinical data with CAR macrophages |
| Mediates phagocytosis and cytotoxicity | Numerically few necessitating ex vivo expansion | In non–CAR-engineered cells: | |
| Cross present antigens to αβ T cells | No GVHD | ||
| No toxicities | |||
| CIK | Multiple killing mechanisms including MHC-dependent and MHC-independent | Heterogeneous products | No clinical data with CAR CIK |
| Numerically few necessitating ex vivo expansion | In non–CAR-engineered cells: | ||
| Lower GVHD risk than T cells60 |
CRISPR-mediated gene editing as the next frontier for cell therapies
Will a bacterial and archaeal immune system adapted for eukaryotic gene editing further elevate the field of cell therapy? Genome editing technologies allow researchers to modify the genome by adding, removing, or otherwise altering the DNA. Several approaches have been devised, including zinc fingers, transcription activator–like effector nucleases, and most recently the clustered regularly interspaced short palindromic repeats (CRISPR) system.46 The discovery of CRISPR revolutionized the field of gene editing because of its simplicity, efficiency, reproducibility, and low cost.47 The CRISPR-Cas system is an RNA-mediated bacterial defense system against viruses (bacteriophages) and plasmids that was repurposed for precise RNA-programmable genome editing in mammalian cells.48
Briefly, the CRISPR-Cas9 technology used in the laboratory for gene editing relies on 2 key elements: the Cas9 enzyme, which acts as a pair of molecular scissors to cut DNA at a specific locus; and a piece of RNA, called guide RNA, which consists of 2 fragments (the trans-activating CRISPR RNA, which binds to the Cas9 enzyme, and the CRISPR RNA, an 18- to 20-nucleotide sequence that is predesigned to recognize a complementary DNA target site in a gene of interest). The guide RNA can therefore guide the Cas9 enzyme to the desired target sequence for gene editing.49,50 The CRISPR-Cas9 tool can also be used to target multiple genes simultaneously by using multiple single-guide RNAs.51
It is important to note that nonspecific and unintended genetic modifications such as insertions or deletions at off-target cleavage sites can arise through the use of engineered nuclease technologies such as CRISPR gene editing.52 For clinical applications, identification of even low-frequency alterations will be critically important. Thus, careful evaluation of off-target effects via technologies such as GUIDE-Seq,53 CIRCLE-Seq,54 and rhampSeq55 is essential before CRISPR-based therapies can be translated to the clinic. The use of ribonucleoprotein complexes and high-fidelity Cas9 was recently shown to significantly reduce the occurrence of such unwanted DNA changes.56
Given the versatility of this gene editing technology, one can imagine its potential applications in cell therapy. A good starting point would be to modify the function of immune effector cells by selectively suppressing negative regulators of cytolytic activity and increasing activation signals. This technology can also be used to fine-tune the safety of cellular therapy products by targeting genes associated with toxicity.57 Positive steps in CRISPR-modified adoptive cell therapy in cancer were recently reported by Stadtmauer et al.58 In this first-in-human pilot study, the investigators isolated autologous T cells from the blood of patients with refractory cancer and electroporated them with CRISPR-Cas9 ribonucleoprotein complexes targeting TRAC, TRBC1, and TRBC2 to suppress the endogenous TCR and PDCD1 to reduce programmed cell death protein 1 expression. The cells were then transduced with a lentiviral vector to express a TCR specific for the cancer–testis antigens NY-ESO-1 and LAGE-1, ex vivo expanded, and then returned to the patients via intravenous infusion.58 This phase 1 study established the feasibility and initial safety of multiplex CRISPR-Cas9–mediated genome engineering of human T-cells. Other clinical trials evaluating CRISPR-modified adoptive cell therapy are under way, as summarized in Table 3. Our group has developed a method to combine CAR engineering with CRISPR-Cas9 gene editing in primary NK cells,59 and we are working on developing a good manufacturing practice–compliant strategy for the production of off-the-shelf CRISPR-modified cord blood–derived CAR-NK cells for the treatment of patients with cancer (visual abstract, panel B).
CRISPR-Cas modified cell therapies for cancer
| National Clinical Trial identifier | Clinical trial phase | Cancer type | CRISPR target | Cell source | Method of CRISPR | Other cell engineering | Status | Location |
|---|---|---|---|---|---|---|---|---|
| NCT04037566 | 1 | Relapsed or refractory ALL and B-cell lymphoma | HPK1 | Autologous T cells | Electroporation of RNP | CD19 CAR lentiviral vector | Recruiting | China |
| NCT03399448 | 1 | Multiple myeloma | Endogenous TCRα, TCRβ, and PD-1 | Autologous T cells | Electroporation of RNP | NYESO-1 TCR lentiviral vector | Active, not recruiting | University of Pennsylvania |
| Melanoma | ||||||||
| Synovial sarcoma | ||||||||
| Myxoid/round cell liposarcoma | ||||||||
| NCT03164135 | 1 | Hematologic malignancies with HIV infection | CCR5 | CD34+ hematopoietic stem/progenitor cells | Unknown | None | Recruiting | China |
| NCT03545815 | 1 | Mesothelin positive | Endogenous TCR and PD-1 | T cells, unknown source | Unknown | Anti-mesothelin CAR | Recruiting | China |
| Solid tumors | ||||||||
| NCT04244656 | 1 | Refractory multiple myeloma | TCR and β2 M gene | Allogeneic T cells | Unknown | Anti-BCMA CAR insertion at the TRAC locus | Recruiting | USA (Nashville and Oregon) and Australia |
| NCT03747965 | 1 | Mesothelin positive | PD-1 | T cells, unknown source | Unknown | Anti-mesothelin CAR | Recruiting | China |
| Solid tumors | ||||||||
| NCT04035434 | 1/2 | B-cell malignancies | TCR and β2 M gene | Allogeneic T cells | Unknown | Anti-CD19 CAR insertion at the TRAC locus | Recruiting | Multiple sites in USA and Australia |
| NCT03166878 | 1/2 | B-cell leukemia and lymphoma | TCR and β2 M gene | Allogeneic T cells from healthy unrelated donors | Electroporation of RNP | Anti-CD19 CAR, lentiviral vector 41BB-CD3ζ | Recruiting | China |
| NCT03044743 | 1/2 | EBV associated malignancies | PD-1 | EBV CTL from autologous source | Unknown | None | Recruiting | China |
| National Clinical Trial identifier | Clinical trial phase | Cancer type | CRISPR target | Cell source | Method of CRISPR | Other cell engineering | Status | Location |
|---|---|---|---|---|---|---|---|---|
| NCT04037566 | 1 | Relapsed or refractory ALL and B-cell lymphoma | HPK1 | Autologous T cells | Electroporation of RNP | CD19 CAR lentiviral vector | Recruiting | China |
| NCT03399448 | 1 | Multiple myeloma | Endogenous TCRα, TCRβ, and PD-1 | Autologous T cells | Electroporation of RNP | NYESO-1 TCR lentiviral vector | Active, not recruiting | University of Pennsylvania |
| Melanoma | ||||||||
| Synovial sarcoma | ||||||||
| Myxoid/round cell liposarcoma | ||||||||
| NCT03164135 | 1 | Hematologic malignancies with HIV infection | CCR5 | CD34+ hematopoietic stem/progenitor cells | Unknown | None | Recruiting | China |
| NCT03545815 | 1 | Mesothelin positive | Endogenous TCR and PD-1 | T cells, unknown source | Unknown | Anti-mesothelin CAR | Recruiting | China |
| Solid tumors | ||||||||
| NCT04244656 | 1 | Refractory multiple myeloma | TCR and β2 M gene | Allogeneic T cells | Unknown | Anti-BCMA CAR insertion at the TRAC locus | Recruiting | USA (Nashville and Oregon) and Australia |
| NCT03747965 | 1 | Mesothelin positive | PD-1 | T cells, unknown source | Unknown | Anti-mesothelin CAR | Recruiting | China |
| Solid tumors | ||||||||
| NCT04035434 | 1/2 | B-cell malignancies | TCR and β2 M gene | Allogeneic T cells | Unknown | Anti-CD19 CAR insertion at the TRAC locus | Recruiting | Multiple sites in USA and Australia |
| NCT03166878 | 1/2 | B-cell leukemia and lymphoma | TCR and β2 M gene | Allogeneic T cells from healthy unrelated donors | Electroporation of RNP | Anti-CD19 CAR, lentiviral vector 41BB-CD3ζ | Recruiting | China |
| NCT03044743 | 1/2 | EBV associated malignancies | PD-1 | EBV CTL from autologous source | Unknown | None | Recruiting | China |
BCMA, B-cell maturation antigen; EBV, Epstein–Barr virus; HPK1, hematopoietic progenitor kinase 1; NYESO-1, New York esophageal squamous cell carcinoma 1; RNP, ribonucleoprotein; TRAC, T-cell receptor-α constant.
Conclusions
CAR-T cell therapy has emerged from an exciting concept at the beginning of this century to a highly effective treatment with curative potential in B-cell malignancies. Nonetheless, despite its many advantages over other forms of cancer therapy, including in vivo expansion and long-term persistence, treatment with CAR-T cells remains a work in progress. Current limitations of this therapy are being overcome by the introduction of alternative platforms for CAR engineering, including NK cells, and the testing of innovative methods such as CRISPR-Cas9 gene editing to counteract tumor-initiated immunosuppressive tactics. If these efforts are successful, we can look forward to a time when clinical applications of cell therapies for cancer are routine rather than investigational strategies at the margins of frontline treatment.
Data are available from the corresponding author, Katayoun Rezvani (KRezvani@mdanderson.org).
This article was selected by the Blood Advances and Hematology 2020 American Society of Hematology Education Program editors for concurrent submission to Blood Advances and Hematology 2020. It is reprinted in Hematology Am Soc Hematol Educ Program. 2020, volume 2020.
Acknowledgments
This work was supported in part by the generous philanthropic contribution to The University of Texas MD Anderson Cancer Center Moonshots Program by grants from CPRIT (RP160693), the Leukemia & Lymphoma Society (6555-18), and Stand Up to Cancer (award SU2C-AACR-DT29-19); by grants (1 R01 CA211044-01, 5 P01CA148600-03, and P50CA100632-16) from the National Institutes of Health (NIH); and by a grant (CA016672) to the MD Anderson Cancer Center from the NIH.
Authorship
Contribution: R.B., M.D., and K.R. wrote and approved the review article.
Conflict-of-interest disclosure: K.R., M.D., R.B., and The University of Texas MD Anderson Cancer Center (MDACC) have an institutional financial conflict of interest with Takeda Pharmaceutical for the licensing of the technology related to the research mentioned here. MD Anderson has implemented an Institutional Conflict of Interest Management and Monitoring Plan to manage and monitor the conflict of interest with respect to MDACC’s conduct of any other ongoing or future research related to this relationship. Off-label drug use: None disclosed.
Correspondence: Katayoun Rezvani, Department of Stem Cell Transplantation and Cellular Therapy, The University of Texas MD Anderson Cancer Center, Houston, TX 77030-4009; e-mail: KRezvani@mdanderson.org.

