

Key Points
By disrupting basal transcription machinery, HBZ RNA inhibits sense transcription of human T-cell leukemia virus type 1.
Repression of genomic expression may allow entry into proviral latency and escape from immune response.

Introduction
At least 20 million people worldwide are estimated to be infected with the oncogenic retrovirus, human T-cell leukemia virus (HTLV) type 1 (HTLV-1).1 HTLV infection is common in southwestern Japan, sub-Saharan Africa, the Caribbean islands, some regions of South America, the Middle East, and Austro-Melanesia. Recent prevalence data from central Australia indicate that up to 45% of adults live with HTLV-1.2 Infected individuals are at risk of developing a rapidly progressive malignancy, adult T-cell leukemia (ATL), and a debilitating and sometimes fatal neurologic condition, HTLV-I–associated myelopathy/tropical spastic paraparesis.3
According to the current most accepted model, 2 viral proteins (TAX and HBZ) are hypothesized to have the highest impact on viral replication and cell transformation.4 The modes of action of TAX and HBZ are remarkably pleiotropic and involve a variety of cell-signaling pathways (CREB, NF-κB, AP1, and AKT). Experimental evidence further shows that TAX drives tumor formation in transgenic mouse models, supporting its oncogenic potential.5 In ∼50% of ATL cases, TAX is either inactivated by genetic mutation or transcriptionally silenced by hypermethylation or deletion of the 5′ long terminal repeat (5′LTR).6 In comparison, HBZ is constitutively transcribed throughout HTLV-1 infection, counteracts TAX-activated viral and cellular pathways, and stimulates cell proliferation.7,8 In HBZ-transgenic models, mice develop systemic inflammation and lymphoma associated with increased effector/memory CD4+ cells and functionally impaired CD4+Foxp3+ regulatory T cells.9 This simplified model thus hypothesizes that TAX initiates transformation whereas HBZ is required to maintain the transformed phenotype providing that TAX expression is silenced.
Besides a role of the TAX and HBZ proteins in driving transformation and promoting viral persistence, more recent evidence has challenged this model. First, the HBZ protein is expressed at very low levels in infected cells.10 Second, the HBZ RNA is mainly localized in the nucleus, consistent with a low rate of translation.11-13 Although the function of the HBZ protein has been clearly evidenced, the dominant nuclear localization of the HBZ RNA thus suggests other regulatory roles such as, for example, epigenetic modulation of gene expression. And, indeed, an untranslatable HBZ RNA devoid of an initiation codon induces the proliferation of a human interleukin 2–dependent T-cell line.14
In this context, we were interested in the functions specifically associated with the HBZ RNA.
Methods
Luciferase reporter assays
The effect of HBZ RNA was evaluated by luciferase reporter assays as described in supplemental Methods.
RTqPCR
Sense and antisense transcription of viral genes was quantified by reverse transcription quantitative polymerase chain reaction (RT-qPCR) as described in supplemental Methods.
Chromatin immunoprecipitation
Interaction of LTR chromatin with RNA polymerase II (RNAPII), TATA box–binding protein (TBP), CREB, and phosphorylated CREB (p-CREB) was performed as described in supplemental Methods.
Chromatin isolation by RNA purification (qPCR)
Fragmented chromatin was precipitated with biotinylated probes complementary to HBZ RNA as described.15 LTR DNA was quantified by qPCR with the primers in supplemental Table 1.
Further details on cell lines, reagents, and protocols are described in supplemental Methods.
Results and discussion
To dissociate RNA and protein-associated functions, we used an expression vector encoding HBZ RNA unable to express the HBZ polypeptide (ie, ATG initiation codon mutated in TTG) (Figure 1A). Upon transfection of this vector into human HEK293 cells, expression of the HBZ protein was absent. In contrast, vectors encoding wild-type HBZ or SM (a codon-matched RNA-scrambled sequence) revealed HBZ protein expression (Figure 1B). To evaluate the effect of HBZ RNA on LTR promoter activity, luciferase-based reporter assays were performed in HEK293 cells containing an integrated LTR promoter. Transfection of HBZ, TTG, or SM vectors affected basal promoter activity (Figure 1C). All 3 vectors also significantly reduced TAX-induced transactivation of the LTR (Figure 1D). Inhibition of TAX-directed transactivation was associated with the presence of HBZ RNA (Figure 1E) and similar amounts of TAX protein (Figure 1F). These data thus confirm that the HBZ protein inhibits LTR-directed promoter activity as reported in the literature.16-18 Note that HBZ SM messenger RNA cannot be detected with the primers as the sequence is altered (Figure 1E). RT-qPCR using SM-specific primers demonstrated that this mutant was correctly expressed upon transduction (data not shown). Results further show that the HBZ RNA in the absence of HBZ protein inhibits the TAX transactivation activity of an integrated LTR promoter. This inhibitory phenotype is not specific to human cells as illustrated by another model in CHOK1 fibroblasts (supplemental Figure 1). In contrast, unspliced HBZ or a cellular noncoding RNA (HOTAIR) does not reduce Tax-dependent transactivation of the LTR (supplemental Figure 1E-F).
![HBZ RNA inhibits basal and TAX-dependent transactivation of the 5′LTR promoter and decreases sense transcription in HTLV-1–infected C91PL lymphocytes. (A) Schematic representation of wild-type HBZ and mutants (TTG and SM) inserted into pME18Sneo vectors. (B) HEK293 cells were transfected using polyethylenimine (PEI) with 2 µg of vectors encoding TAX (pSGTax) and HBZ (as displayed in panel A). After 48 hours, cell lysates were analyzed by immunoblot using anti-TAX and anti-HBZ antibodies. Tubulin was used as loading control. (C) HEK-LTRLuc cells containing a stably integrated pLTRLuc plasmid were transfected with 1 µg of pME18SNeo (vector), pME18SNeo-HBZ, pME18SNeo-TTG, or pME18SNeo-SM. The firefly luciferase activities (light arbitrary units) were measured at 48 hours posttransfection. (D) HEK-LTRLuc cells were transfected with pSG5, pSGTax, pME18SNeo, pME18SNeo-HBZ, pME18SNeo-SM, and pME18SNeo-TTG vectors (1 µg). (E) At 48 hours, the levels of HBZ RNA were quantified by RT-qPCR using HBZ1 primers (supplemental Table 1) and normalized to those of the HPRT housekeeping gene. (F) In parallel, TAX expression was determined by immunoblot as described in panel B. Numbers indicate the mean quantification of band luminescence intensities of TAX relative to tubulin resulting from 3 independent immunoblots. (G-I) HTLV-1 immortalized lymphocytes C91PL (not transduced [NT]) were stably transduced with pUCOE-SFFV-EGFP, pUCOE-SFFV-HBZ, pUCOE-SFFV-TTG, and pUCOE-SFFV-SM lentiviruses expressing mock (enhanced green fluorescent protein [EGFP]), HBZ TTG or SM, respectively. The leukemic HTLV-1–infected ATL2 cell line was analyzed in parallel as reference for physiological HBZ RNA levels. HBZ (G), TAX (H), and GAG (I) RNA levels were quantified by RT-qPCR and normalized to HPRT. All data are presented as mean plus or minus standard error of at least 3 independent experiments. Statistical P values were calculated according to the Tukey multiple comparison tests.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/21/10.1182_bloodadvances.2020001675/3/m_advancesadv2020001675f1.png?Expires=1767713259&Signature=4RDWWqgEc-YJUbtcdClgRkNIsuU1TlGeS3UOglcyZR3EKmF5JpldL~nTRcnSzbvUjUzIm73fMdj5AC5q0YyWHGpUvFEcO9SYqh5xnUg86j-6Fbur9MPz0OFPp4ZjmWux2nQJjvnOKpWwGgddmVrvu1M4vkbbZGgxoGpImGwOmL25~d5XiMRgW2kZ1Igzc6ELvYJmLsp3bGBwr8m3qYNPA53bZjv0LEyFaSaVi3-8pXq8B5FPEGzcjMCNnay0ODif8UokKjVoJNFaOOLCHhrwX2m0iRxe9ghZly2ouvdfwO6aQx~XnXNQdWOihjFcW7FsiU2Q5Iq~TgjjjSkzu-1uzQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
HBZ RNA inhibits basal and TAX-dependent transactivation of the 5′LTR promoter and decreases sense transcription in HTLV-1–infected C91PL lymphocytes. (A) Schematic representation of wild-type HBZ and mutants (TTG and SM) inserted into pME18Sneo vectors. (B) HEK293 cells were transfected using polyethylenimine (PEI) with 2 µg of vectors encoding TAX (pSGTax) and HBZ (as displayed in panel A). After 48 hours, cell lysates were analyzed by immunoblot using anti-TAX and anti-HBZ antibodies. Tubulin was used as loading control. (C) HEK-LTRLuc cells containing a stably integrated pLTRLuc plasmid were transfected with 1 µg of pME18SNeo (vector), pME18SNeo-HBZ, pME18SNeo-TTG, or pME18SNeo-SM. The firefly luciferase activities (light arbitrary units) were measured at 48 hours posttransfection. (D) HEK-LTRLuc cells were transfected with pSG5, pSGTax, pME18SNeo, pME18SNeo-HBZ, pME18SNeo-SM, and pME18SNeo-TTG vectors (1 µg). (E) At 48 hours, the levels of HBZ RNA were quantified by RT-qPCR using HBZ1 primers (supplemental Table 1) and normalized to those of the HPRT housekeeping gene. (F) In parallel, TAX expression was determined by immunoblot as described in panel B. Numbers indicate the mean quantification of band luminescence intensities of TAX relative to tubulin resulting from 3 independent immunoblots. (G-I) HTLV-1 immortalized lymphocytes C91PL (not transduced [NT]) were stably transduced with pUCOE-SFFV-EGFP, pUCOE-SFFV-HBZ, pUCOE-SFFV-TTG, and pUCOE-SFFV-SM lentiviruses expressing mock (enhanced green fluorescent protein [EGFP]), HBZ TTG or SM, respectively. The leukemic HTLV-1–infected ATL2 cell line was analyzed in parallel as reference for physiological HBZ RNA levels. HBZ (G), TAX (H), and GAG (I) RNA levels were quantified by RT-qPCR and normalized to HPRT. All data are presented as mean plus or minus standard error of at least 3 independent experiments. Statistical P values were calculated according to the Tukey multiple comparison tests.
HBZ RNA inhibits basal and TAX-dependent transactivation of the 5′LTR promoter and decreases sense transcription in HTLV-1–infected C91PL lymphocytes. (A) Schematic representation of wild-type HBZ and mutants (TTG and SM) inserted into pME18Sneo vectors. (B) HEK293 cells were transfected using polyethylenimine (PEI) with 2 µg of vectors encoding TAX (pSGTax) and HBZ (as displayed in panel A). After 48 hours, cell lysates were analyzed by immunoblot using anti-TAX and anti-HBZ antibodies. Tubulin was used as loading control. (C) HEK-LTRLuc cells containing a stably integrated pLTRLuc plasmid were transfected with 1 µg of pME18SNeo (vector), pME18SNeo-HBZ, pME18SNeo-TTG, or pME18SNeo-SM. The firefly luciferase activities (light arbitrary units) were measured at 48 hours posttransfection. (D) HEK-LTRLuc cells were transfected with pSG5, pSGTax, pME18SNeo, pME18SNeo-HBZ, pME18SNeo-SM, and pME18SNeo-TTG vectors (1 µg). (E) At 48 hours, the levels of HBZ RNA were quantified by RT-qPCR using HBZ1 primers (supplemental Table 1) and normalized to those of the HPRT housekeeping gene. (F) In parallel, TAX expression was determined by immunoblot as described in panel B. Numbers indicate the mean quantification of band luminescence intensities of TAX relative to tubulin resulting from 3 independent immunoblots. (G-I) HTLV-1 immortalized lymphocytes C91PL (not transduced [NT]) were stably transduced with pUCOE-SFFV-EGFP, pUCOE-SFFV-HBZ, pUCOE-SFFV-TTG, and pUCOE-SFFV-SM lentiviruses expressing mock (enhanced green fluorescent protein [EGFP]), HBZ TTG or SM, respectively. The leukemic HTLV-1–infected ATL2 cell line was analyzed in parallel as reference for physiological HBZ RNA levels. HBZ (G), TAX (H), and GAG (I) RNA levels were quantified by RT-qPCR and normalized to HPRT. All data are presented as mean plus or minus standard error of at least 3 independent experiments. Statistical P values were calculated according to the Tukey multiple comparison tests.
To investigate the biological relevance of the phenotype in HTLV-infected cells, C91PL lymphocytes reported to express low levels of HBZ RNA19 were transduced with retroviral vectors encoding either HBZ, TTG, or SM (Figure 1G). The levels of HBZ RNA upon transduction were similar to those measured in patient-derived ATL2 cells. In these conditions, the levels of 2 sense RNAs (TAX and GAG/POL) were significantly reduced (Figure 1H-I). The same conclusion was drawn in HUT102 HTLV-infected lymphocytes (supplemental Figure 2).
Together, these data demonstrate that, at physiologically relevant levels, HBZ RNA inhibits 5′LTR–directed sense transcription in HTLV-infected lymphocytes.
To characterize the mechanisms involved, the interaction of HBZ RNA with the LTR promoter was evaluated by chromatin isolation by RNA purification (ChIRP; Figure 2A). Using a set of 32 biotinylated probes, HBZ RNA was retrieved from TTG-transduced CHOK1 cells (Figure 2B). As control, only background levels of HBZ RNA were precipitated with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) probes. In these conditions, LTR chromatin was specifically recovered upon expression of the TTG RNA into CHOK1 cells (Figure 2C). Interaction of HBZ RNA was further validated in HTLV-infected lymphocytes (C8166 and MT4) but not in control Jurkat cells (Figure 2D-E).
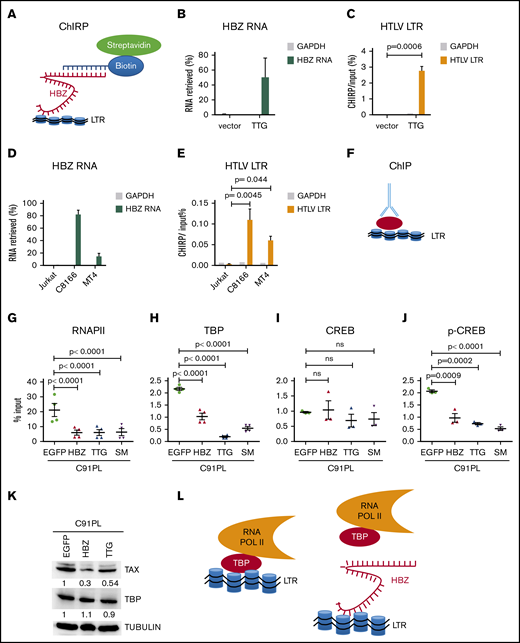
HBZ RNA inhibits interaction of TBP with LTR chromatin. (A) Schematic representation of ChIRP. Biotinylated probes (supplemental Table 2) were hybridized to target HBZ RNA, and chromatin complexes were purified using magnetic streptavidin beads. (B) Enrichment of HBZ RNA in ChIRP precipitates from CHOK1-LTRLuc cells transfected with pME18Sneo or pME18Sneo-TTG plasmids. Data are percentages of HBZ and GAPDH RNAs retrieved relatively to the input DNA from RNA fractions. (C) LTR chromatin was precipitated with HBZ-specific biotinylated probes. DNA was eluted and amplified by qPCR using GAPDH or HTLV U3-R LTR primers (LTR1; supplemental Table 1). (D) Retrieval of HBZ RNA-specific RNA by ChIRP. The RNA fraction of ChIRP precipitates from control Jurkat T cells and HTLV-1–infected lymphocytes (C8166 and MT4) was extracted and analyzed by RT-qPCR, using primers against GADPH and HBZ RNAs (HBZ2; supplemental Table 1). (E) DNA fraction from ChIRP of Jurkat, C8166, and MT4 was extracted and analyzed by qPCR using primers against HTLV LTR (LTR1) and a control locus (GADPH). (F) Schematic representation of ChIP. (G-H) C91PL lymphocytes transduced with pUCOE-SFFV-EGFP, HBZ, TTG, or SM lentiviruses were analyzed by ChIP using antibodies specific for RNAPII (G), TBP (H), CREB (I), and CREB phosphorylated at serine 133 (p-CREB; J). Data shown are percentages of LTR DNA immunoprecipitated from the input before ChIP. The values are averages and mean standard error of at least 3 independent experiments. (K) C91PL lymphocytes expressing EGFP, HBZ, or TTG (Figure 1) were analyzed by immunoblot using anti-TAX, anti-TBP, and anti-tubulin antibodies. Numbers indicate the mean quantification of band luminescence intensities of TAX and TBP relative to tubulin obtained from 2 independent experiments. Statistical significances were calculated using analysis of variance and the Tukey post hoc test. (L) Model of inhibition of 5′LTR-directed transcription by HBZ RNA.
HBZ RNA inhibits interaction of TBP with LTR chromatin. (A) Schematic representation of ChIRP. Biotinylated probes (supplemental Table 2) were hybridized to target HBZ RNA, and chromatin complexes were purified using magnetic streptavidin beads. (B) Enrichment of HBZ RNA in ChIRP precipitates from CHOK1-LTRLuc cells transfected with pME18Sneo or pME18Sneo-TTG plasmids. Data are percentages of HBZ and GAPDH RNAs retrieved relatively to the input DNA from RNA fractions. (C) LTR chromatin was precipitated with HBZ-specific biotinylated probes. DNA was eluted and amplified by qPCR using GAPDH or HTLV U3-R LTR primers (LTR1; supplemental Table 1). (D) Retrieval of HBZ RNA-specific RNA by ChIRP. The RNA fraction of ChIRP precipitates from control Jurkat T cells and HTLV-1–infected lymphocytes (C8166 and MT4) was extracted and analyzed by RT-qPCR, using primers against GADPH and HBZ RNAs (HBZ2; supplemental Table 1). (E) DNA fraction from ChIRP of Jurkat, C8166, and MT4 was extracted and analyzed by qPCR using primers against HTLV LTR (LTR1) and a control locus (GADPH). (F) Schematic representation of ChIP. (G-H) C91PL lymphocytes transduced with pUCOE-SFFV-EGFP, HBZ, TTG, or SM lentiviruses were analyzed by ChIP using antibodies specific for RNAPII (G), TBP (H), CREB (I), and CREB phosphorylated at serine 133 (p-CREB; J). Data shown are percentages of LTR DNA immunoprecipitated from the input before ChIP. The values are averages and mean standard error of at least 3 independent experiments. (K) C91PL lymphocytes expressing EGFP, HBZ, or TTG (Figure 1) were analyzed by immunoblot using anti-TAX, anti-TBP, and anti-tubulin antibodies. Numbers indicate the mean quantification of band luminescence intensities of TAX and TBP relative to tubulin obtained from 2 independent experiments. Statistical significances were calculated using analysis of variance and the Tukey post hoc test. (L) Model of inhibition of 5′LTR-directed transcription by HBZ RNA.
Because interaction of HBZ RNA with the LTR promoter potentially interferes with the transcription machinery, chromatin immunoprecipitation (ChIP) assays were performed with antibodies specific to the TBP and RNAPII (Figure 2F). In presence of HBZ RNA, interaction of RNAPII with LTR chromatin was reduced in CHOK1 cells (supplemental Figure 3) and in C91PL lymphocytes (Figure 2G). ChIP of TBP was also significantly affected in the presence of TTG (supplemental Figure 3; Figure 2H). Concomitantly, less CREB phosphorylated at serine 133 was bound to the LTR (Figure 2I-J; supplemental Figure 3). In these conditions, HBZ RNA reduced the total amount of Tax protein but did not affect TBP (Figure 2K).
Together, these data highlight the specific interaction of the HBZ messenger RNA with the HTLV-1 promoter in a chromatin context. ChIP further demonstrates that the HBZ RNA directly affects interaction of RNAPII with the LTR promoter by displacing TBP, the initiator of basal transcription (Figure 2L). This mechanism impairs 5′LTR-directed gene expression of structural, enzymatic, and regulatory proteins. Because these viral proteins stimulate a vigorous cytotoxic and humoral immune response in HTLV-1–infected individuals, HBZ RNA-dependent inhibition of sense transcription could be a key mechanism required for persistence of infected cells. Although p30, p13, and HBZ are known to suppress TAX-dependent transactivation from the viral promoter,4,20 a remaining question is how silencing of proviral expression is achieved or maintained in the absence of viral proteins. Nuclear retention of HBZ RNA would prevent expression of the HBZ protein, which stimulates an efficient cytotoxic cell-mediated immune response.21 This report describes a novel mechanism by which the HBZ RNA in the absence of HBZ protein inhibits sense transcription at physiologically relevant levels in lymphocytes derived from HTLV-1–infected patients. Because HBZ RNA also inhibits basal transcription of the 5′LTR promoter in the absence of Tax, this mechanism may lead to a complete silencing of viral expression, as observed in single-cell imaging.22
In conclusion, we have unraveled an RNA-dependent epigenetic mechanism by which the HTLV-1 provirus undergoes shutdown of sense transcription, thereby allowing entry into latency and escape from immune response. Inhibition of this mechanism opens new perspectives for potential clinical applications for HTLV-1–associated diseases.
Data-sharing requests may be e-mailed to the corresponding author, Luc Willems, at luc.willems@uliege.be.
Acknowledgments
The authors thank the GIGA technological platforms for support. The authors are grateful to Christelle Gillissen, Nathalie Renotte, and Jean-Rock Jacques for technical expertise. The authors are also grateful to Françoise Bex (University of Brussels, Brussels, Belgium), Emmanuel Di Valentin (GIGA Viral Vectors platform), Patrick Green (The Ohio State University, Columbus, OH), Masao Matsuoka (Kumamoto and Kyoto Universities, Kumamoto and Kyoto, Japan), and Jean-Michel Mesnard and Jean-Marie Peloponese (University of Montpellier, Montpelier, France) for providing reagents (plasmids and antibodies).
This work was supported by the Fonds National de la Recherche Scientifique (FNRS; PDR T.0261.20), Télévie (7.4599.19), the Fonds Spéciaux pour la Recherche of Uliege, the Belgian Foundation against Cancer, and the Fondation Léon Fredericq (FLF). H.G. was supported by fellowships of the Belgian Foundation against Cancer and Télévie. P.S.C. benefits from a COFUND grant and a Belgian Foundation against Cancer fellowship. L.W. is research director of the FNRS.
Authorship
Contribution: H.G. designed protocols, performed experiments, and analyzed data; P.S.C., F.P., G.B.H., and R.A. contributed to experiments and discussed conclusions; L.W. designed the project, analyzed data, and drafted the manuscript; and all authors edited and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Luc Willems, Molecular and Cellular Epigenetics, Interdisciplinary Cluster for Applied Genoproteomics (GIGA) of University of Liège (ULg), Sart Tilman B34, Av de l'Hôpital No. 1, 4000 Liège, Belgium; e-mail: luc.willems@uliege.be.

