

Key Points
Because classic randomized trials are impractical in hemophilia, a Bayesian platform design was used to design 2 linked inhibitor trials.
Simulations indicate the platform design provides statistical and administrative efficiency to run prevention and eradication trials.

Abstract
Among individuals with the rare congenital bleeding disorder hemophilia A, the major challenge is inhibitor formation, which is associated with significant morbidity and cost. Yet, as the optimal approach to prevent and eradicate inhibitors is not known, we are at equipoise. Because classic trial design is not practical in a rare disease setting, we designed 2 48-week randomized trials comparing ELOCTATE and emicizumab to prevent and eradicate inhibitors. To achieve statistical efficiency, we incorporated historic data (Bayesian priors) on inhibitor formation to allow preferential randomization to emicizumab, piecewise exponential survival models to determine mean and 95% confidence interval for inhibitor formation in each arm, and simulations to determine the best model design to optimize power. To achieve administrative efficiency, the trials will be performed with the same sites, staff, visit frequency, blood sampling, laboratories, and laboratory assays, with streamlined enrollment so patients developing inhibitors in the first trial may be enrolled on the second trial. The primary end point is the probability of inhibitor formation or inhibitor eradication, respectively. The design indicates early stopping rules for overwhelming evidence of superiority of the emicizumab arms. Simulations indicate that, with 66 subjects, the Prevention Trial will have 84% power to detect noninferiority of emicizumab to ELOCTATE with a margin of 10% if emicizumab is truly 10% superior to ELOCTATE; with 90 subjects, the Eradication Trial will have 80% power to detect 15% superiority of ELOCTATE immune tolerance induction with vs without emicizumab. Thus, a platform design provides statistical and administrative efficiency to conduct INHIBIT trials.
Introduction
Hemophilia A is an X-linked congenital bleeding disorder, occurring 1 in 5000 male births, that results from deficiency of factor VIII (FVIII) and is characterized by spontaneous and traumatic bleeding into joints, muscles, and body cavities. Among the most serious complications of the disease is inhibitor formation, which affects 25% of those with severe hemophilia A. Inhibitor formation is a T-cell–dependent B-cell–mediated immune response to exogenous FVIII,1-3 which renders lifesaving FVIII ineffective and leads to poorly controlled bleeding, frequent hospitalizations, and high health care costs.4-6 Thus, prevention and eradication of inhibitors are major goals of hemophilia treatment,7 which, if successful, would be adopted into clinical practice.
Despite the availability of 2 US Food and Drug Administration–approved novel hemostatic agents, specifically ELOCTATE, an Fc-fusion protein with extended half-life that prevents and treats bleeds in children and adults with hemophilia A,8-11 and emicizumab, a bispecific monoclonal antibody FVIII mimetic that prevents bleeds in children and adults with hemophilia A with or without inhibitors,12-16 the optimal approach to inhibitor prevention and inhibitor eradication is unknown. Given the state of equipoise regarding inhibitor prevention and inhibitor eradication, there is impetus to conduct clinical trials comparing these novel agents to determine the optimal approach to preventing and eradicating hemophilia inhibitors. The basis for equipoise and motive behind each trial are summarized below.
In the Inhibitor Prevention Trial, we propose to determine whether a treatment strategy incorporating emicizumab is noninferior to ELOCTATE in inhibitor prevention in previously untreated patients (PUPs). The motive behind this trial is that, although ELOCTATE induces regulatory T cells via Fc that may promote tolerance to FVIII,8,9 inhibitors still develop in 25% of PUPs.17-19 Although emicizumab reduces bleeds, bleed intensity, and FVIII exposure (each are risks for inhibitor formation), breakthrough bleeds do occur.14,16 Yet, few long-term data are available in PUPs. Thus, it is unknown whether a treatment strategy incorporating emicizumab reduces bleed intensity and FVIII use sufficiently to avoid activation of the immune system and prevent inhibitor formation, or, whether it simply delays inhibitor formation. Further, until PUPs experience 10 to 20 FVIII exposures during emicizumab prophylaxis, the risk for inhibitor formation may not be known. Thus, continued monitoring for inhibitor formation after trial completion will be necessary to answer this question.
In the Inhibitor Eradication Trial, we propose to determine whether a strategy incorporating ELOCTATE immune tolerance induction (ITI) plus emicizumab is superior to ELOCTATE ITI alone in inhibitor eradication. The motive behind this trial is that, although small uncontrolled observational studies indicate that ELOCTATE may shorten inhibitor eradication20,21 (ie, ITI), few data are available regarding whether a treatment strategy incorporating emicizumab shortens ITI.22 If emicizumab provides better bleed protection in inhibitor patients during ELOCTATE ITI than does ELOCTATE ITI alone, then immune activation may be reduced and fewer inhibitor spikes may occur that delay achieving tolerization and inhibitor eradication. There is precedent for better bleed protection with high-dose vs low-dose FVIII ITI, even in the presence of inhibitors23 ; these findings suggest that FVIII neutralization by the inhibitor does not prevent transient thrombin generation in the absence or transient presence of functional FVIII.24 Because bleed intensity is a significant and separate risk for inhibitors,25 we hypothesize that fewer bleeds will occur with a strategy incorporating the concurrent use of ELOCTATE ITI and emicizumab compared with ELOCTATE ITI alone, and, thereby, shorten the time to inhibitor eradication.
To conduct trials to prevent and eradicate inhibitor formation is challenging because classic trial design is not practical in this rare disease: the population is small, outcomes are rare, and competing studies exist.7,26-29 Standard classic designs based on sample sizes that are feasible result in insufficient power to detect a reasonable effect size. For example, using a standard comparison of proportions with 33 subjects per group provides only 23.9% power to detect noninferiority, with a 10% noninferiority margin when the probability of preventing inhibitor development for each treatment is 85%. If the comparator actually reduces the rate of inhibitor development by 5% to 10%, then power for noninferiority increases to 42.4% or 65.9%, which remains underpowered for a definitive trial.
Approaches to clinical trial design in rare disease settings have been proposed, including using networks of care, relaxed statistical error rates, historical data, carefully selected outcome measures, clinical trial platforms, and Bayesian designs.30-35 The use of Bayesian platform trial design will provide statistical and administrative efficiency for the conduct of the Inhibitor Prevention Trial and the Inhibitor Eradication Trial. Statistical efficiency will be achieved by the (1) use of Bayesian prior distributions to incorporate historical data to increase power and promote efficient use of rare data, (2) use of piecewise exponential survival models to determine mean and 95% confidence interval for each trial arm, and (3) use of thousands of simulations to determine the best model design to optimize power. Administrative efficiency will be achieved by (1) linked trials, with the same sites and with the same staff and visit frequency; (2) uniform procedures, with the same blood sampling, laboratories, laboratory preparation, and assays; and (3) streamlined enrollment, with patients developing inhibitors on the first trial enrolling on the second trial. To this end, we worked with Berry Consultants, a statistical consulting company, through National Institutes of Health National Heart, Lung, and Blood Institute grant X01 HL143024, to develop the INHIBIT Clinical Trials Platform, which is composed of 2 linked phase 3 randomized trials: the Inhibitor Prevention Trial and the Inhibitor Eradication Trial (Figure 1).
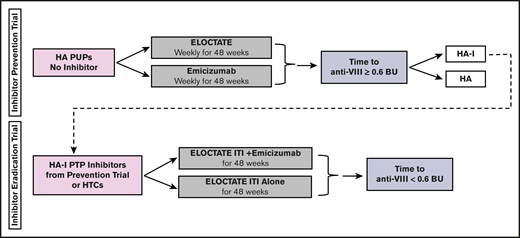
INHIBIT trials Bayesian platform design. Proposed schema for the INHIBIT Trials Platform, which links 2 phase 3 randomized trials to prevent and eradicate inhibitors in individuals with hemophilia: the Inhibitor Prevention Trial and the Inhibitor Eradication Trial. BU, Bethesda units; HA, hemophilia A; HA-I, hemophilia A with inhibitors; HTC, hemophilia treatment center; PTP, previously treated patient.
INHIBIT trials Bayesian platform design. Proposed schema for the INHIBIT Trials Platform, which links 2 phase 3 randomized trials to prevent and eradicate inhibitors in individuals with hemophilia: the Inhibitor Prevention Trial and the Inhibitor Eradication Trial. BU, Bethesda units; HA, hemophilia A; HA-I, hemophilia A with inhibitors; HTC, hemophilia treatment center; PTP, previously treated patient.
Methods
To design this Bayesian platform trial, we performed 2 analyses: an analysis to create the study design and an analysis of the actual study data to be accrued. To maximize the use of available data and power to conduct these trials, we used (1) a platform of 2 linked trials to capitalize on the overlap of patients participating in the prevention and eradication research efforts, (2) Bayesian methodology to formally incorporate existing available information into the analysis, (3) advanced modeling to incorporate time-to-event modeling for the estimation of the binary outcome of primary interest, (4) statistical methods to increase power, and (5) simulations to determine the best model design to optimize power. We also conducted a feasibility study through National Institutes of Health grant U34 HL114674,19 as well as a subsequent informal survey, to ensure that the trial would be consistent with current practice, that patients and physicians would be willing to participate, and that sufficient potential sites and subjects would enroll on the Inhibitor Prevention Trial and the Inhibitor Eradication Trial.
Platform trial design
To more efficiently test the trial hypotheses, we designed a platform trial, the INHIBIT Clinical Trials Platform,32,35 which is composed of 2 linked phase 3 trials: the Inhibitor Prevention Trial and the Inhibitor Eradication Trial (Figure 1). The hypothesis of the Inhibitor Prevention Trial is that emicizumab prophylaxis is noninferior to ELOCTATE prophylaxis in preventing inhibitor development in PUPs with severe hemophilia A when given weekly over 48 weeks (Figure 2). The hypothesis of the Inhibitor Eradication Trial is that ELOCTATE ITI combined with weekly emicizumab prophylaxis is superior to ELOCTATE ITI alone in eradicating inhibitors in previously treated patients (PTPs) with severe hemophilia A and inhibitors over 48 weeks (Figure 3).

Clinical trial 1: Inhibitor Prevention Trial schema. Schema for the Inhibitor Prevention Trial, in which ELOCTATE prophylaxis is compared with emicizumab prophylaxis to reduce inhibitor formation in PUPs with severe hemophilia A. The sample size indicates 1:3 preferential randomization of ELOCTATE/emicizumab, as a result of the incorporation of subjects on the ELOCTATE arm, borrowed from the Bayesian prior (supplemental Methods).
Clinical trial 1: Inhibitor Prevention Trial schema. Schema for the Inhibitor Prevention Trial, in which ELOCTATE prophylaxis is compared with emicizumab prophylaxis to reduce inhibitor formation in PUPs with severe hemophilia A. The sample size indicates 1:3 preferential randomization of ELOCTATE/emicizumab, as a result of the incorporation of subjects on the ELOCTATE arm, borrowed from the Bayesian prior (supplemental Methods).
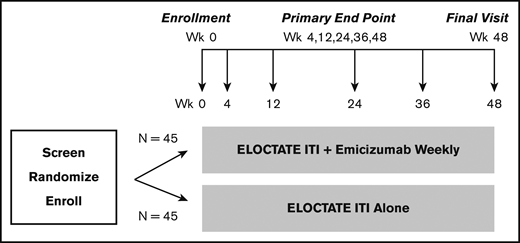
Clinical Trial 2: Inhibitor Eradication Trial schema. Schema for the Inhibitor Eradication Trial, in which ELOCTATE ITI combined with emicizumab prophylaxis is compared with ELOCTATE ITI alone to eradicate inhibitor in PTPs with severe hemophilia A and an inhibitor.
Clinical Trial 2: Inhibitor Eradication Trial schema. Schema for the Inhibitor Eradication Trial, in which ELOCTATE ITI combined with emicizumab prophylaxis is compared with ELOCTATE ITI alone to eradicate inhibitor in PTPs with severe hemophilia A and an inhibitor.
Both trials were constructed for efficiency of design and conducted at the same hemophilia treatment centers (HTCs) by the same staff for data entry and biospecimen tracking, with the same visit frequency, blood collection schema, standardized outcome measures, and the same laboratory and mechanistic assays performed in the same centralized laboratories. In addition, subjects developing inhibitors in the Inhibitor Prevention Trial can be enrolled in the Inhibitor Eradication Trial, along with eligible inhibitor patients at the same HTC, enabling some subjects to participate in both trials.
Bayesian design
Using Bayesian statistical methodologies, known prior information regarding treatments will be formally incorporated into the analysis.30-33 For the Inhibitor Prevention Trial, prior information for the ELOCTATE group was based on data extracted from the open-label single-armed ELOCTATE PUP study.18 Because actual patient-level data were not made available, we recreated the data based on publicly available Kaplan-Meier curves. Specifically, data were generated to replicate a series of survival curves for inhibitor prevention in the 89 PUPs who had received ≥1 exposure of ELOCTATE in that trial. Because there are no known published data on the impact of emicizumab on inhibitor development in PUPs, an estimate with large variance (or noninformative prior) was chosen. For the Inhibitor Eradication Trial, because there are few known published data on the impact of ELOCTATE ITI, with or without emicizumab, on inhibitor eradication in PTPs,20-22 noninformative priors are used for both arms of the trial (see "Development of the prior distributions" in supplemental Methods).
Advanced time-to-event Bayesian modeling
The primary outcomes of the Inhibitor Prevention Trial and the Inhibitor Eradication Trial are the proportion of subjects who develop inhibitors or eliminate inhibitors, respectively, within 48 weeks. These rates will be estimated using survival analysis that incorporates information about the timing of inhibitor development (or eradication), censoring subjects who drop off the trial or who have incomplete data.36,37 This methodology allows interim analyses to make full use of partial data from subjects who have not yet completed the 48-week follow-up window. Based on Kaplan-Meier curves from the PUPs A-LONG study,18 the rate of inhibitor development with ELOCTATE is faster in the first 20 weeks of exposure (∼1 per week) than in weeks 20 to 48.18 Thus, the time to inhibitor development will be modeled as a piecewise exponential distribution, allowing a greater rate of inhibitor development between weeks 0 and 20 than between weeks 20 and 48. Separate models will be fit for each study treatment, resulting in an analysis where the model assumptions are met. From the resulting fitted survival curves, we will estimate the proportion of subjects that develop inhibitors within 48 weeks for each treatment. These proportions will then be compared for noninferiority and superiority in preventing inhibitor formation (see "Bayesian piecewise exponential models" in supplemental Methods). A similar methodology will be implemented for the estimation of the eradication rates.
Randomization allocation
Statistical efficiency in the comparison of 2 treatments is achieved when the sample sizes are balanced between arms. For the Inhibitor Prevention Trial, by using informative prior distributions based on external information on the ELOCTATE arm, we may achieve a similar “effective sample size” with fewer concurrently randomized subjects to that treatment arm, because the analysis will incorporate the concurrent study data and the historical data. Accordingly, we adjust the allocation ratio to shift more of the randomization to the emicizumab arm, the treatment arm with the lesser studied drug and no prior information to incorporate into the model. The advantages of this approach are that (1) many subjects prefer and HTC providers preferentially prescribe emicizumab because of its simpler subcutaneous route, (2) the information gained from emicizumab is maximized, (3) at the conclusion of the trial, a similar proportion of patients will have received ELOCTATE and emicizumab, allowing for a more robust research base in the future, and (4) the power to detect a significant statistical effect, if it exists, is maximized.
Simulations were performed across a range of choices for the randomization ratio, with a resulting decision to use 1:3 for ELOCTATE/emicizumab. With only 66 subjects available for randomization into the Inhibitor Prevention Trial, we aimed to have a minimum of 15 contemporary randomized subjects on the ELOCTATE arm (plus the additional subjects borrowed from the prior) to collect sufficient secondary outcome efficacy and safety data (supplemental Methods). For the Inhibitor Eradication Trial, because there are very few published data regarding the eradication of inhibitors with ELOCTATE ITI, with or without emicizumab, we used noninformative priors and a randomization allocation of 1:1 ELOCTATE ITI plus emicizumab/ELOCTATE ITI alone (see "Power simulations" in supplemental Methods).
Interim data analysis
Within each trial, there will be 1 interim analysis after 75% of the subjects have been randomized. For the Inhibitor Prevention Trial, this will be after the 50th subject is randomized; for the Inhibitor Eradication Trial, this will be after the 68th subject is randomized. All available data for those subjects, including those participating for <48 weeks, will be analyzed according to the amount of information they have contributed through the time-to-event model. For the Inhibitor Prevention Trial, although the primary analysis is a test of noninferiority, the trial will be stopped at the interim if the emicizumab arm demonstrates superiority to the ELOCTATE arm. The Inhibitor Eradication Trial will be stopped early only if the ELOCTATE ITI plus emicizumab arm is different from the ELOCTATE ITI–only arm. The advantage of interim analysis is that it allows either of the trials to stop early if there is overwhelming evidence. However, designs that allow early stopping for trial success must be calibrated appropriately to prevent inflating the probability of drawing false-positive conclusions (see “Power simulations” in supplemental Methods).
Noninferiority and superiority assessments
For the Inhibitor Prevention Trial, if the trial does not stop at the interim, the primary analysis will be for noninferiority (ie, having a high probability [95%] of being no more than 10% worse). If emicizumab is noninferior to ELOCTATE, then the superiority of emicizumab will be tested. This testing provides for the strongest conclusion possible with the data collected. For the Inhibitor Eradication Trial, a 2-sided test for the difference between ELOCTATE ITI plus emicizumab vs ELOCTATE ITI alone will be used. Simulations were performed to determine the specific thresholds for declaring noninferiority or superiority while maintaining a type 1 error rate of 0.05.
Results
Survey projections of sample size
The outcome of the Inhibitor Prevention Trial is the proportion who develop inhibitor formation by 48 weeks. Our 2019 survey indicates that 420 infants with severe hemophilia A will be born in the next 6 years at 31 HTCs.19 If only 16% of these patients participate, we will be able to enroll 66 subjects over the 6-year enrollment period (Table 1). The long time to trial completion is a potential disadvantage of trials in rare disease. The novel therapies available will change, and patient and physician participation may wane accordingly; thus, it is critical to complete the trials as quickly as possible. However, 1 advantage of platform master protocols is the potential to assess new agents as they become available, by adding a new treatment arm and comparing it with the winner of each ongoing trial.7
Survey: projected enrollment on the Inhibitor Prevention Trial and Inhibitor Eradication Trial
| Potential subject pool | Percent needed to enroll in 6-y trial | Sample size and power for 6-y trial |
|---|---|---|
| Inhibitor Prevention Trial | ||
| Severe HA births | ||
| PUPs | ||
| n = 210 in 3 y (2016-2018) | ||
| n = 420 in 6-y trial | 16% of 420 = 66 | n = 66; 1 − β = 0.80, α = 0.05 |
| Inhibitor Eradication Trial | ||
| Severe HA-I | ||
| n = 12 from Prevention Trial plus | ||
| n = 274 ITI-refractory/ITI-naive | 29% of 274 = 78 | n = 90 (12+78); 1 − β = 0.80, α = 0.05 |
| Potential subject pool | Percent needed to enroll in 6-y trial | Sample size and power for 6-y trial |
|---|---|---|
| Inhibitor Prevention Trial | ||
| Severe HA births | ||
| PUPs | ||
| n = 210 in 3 y (2016-2018) | ||
| n = 420 in 6-y trial | 16% of 420 = 66 | n = 66; 1 − β = 0.80, α = 0.05 |
| Inhibitor Eradication Trial | ||
| Severe HA-I | ||
| n = 12 from Prevention Trial plus | ||
| n = 274 ITI-refractory/ITI-naive | 29% of 274 = 78 | n = 90 (12+78); 1 − β = 0.80, α = 0.05 |
The projections are based on a survey of HTCs. The data indicate that the trials are possible if ≥16% of available PUPs with severe hemophilia A (HA) enroll on the Inhibitor Prevention Trial and if ≤29% of PTPs with severe HA with inhibitors (HA-I) enroll on the Inhibitor Eradication Trial.
The outcome of the Inhibitor Eradication Trial is the proportion of subjects who achieve inhibitor eradication by 48 weeks. Our 2019 survey indicates that there are 78 ITI-refractory or ITI-naive inhibitor patients at 31 HTCs. When combined with 12 subjects on the Inhibitor Prevention Trial who are expected to develop inhibitors, this will provide a total of 90 inhibitor subjects (78 + 12) available to enroll over the 6-year enrollment (Table 1). This survey indicates that the Inhibitor Eradication Trial is feasible if ≥29% of available severe hemophilia A patients with inhibitors enroll in the trial.
Control of type 1 error
A type 1 error, represented by an α level in statistical hypothesis testing, is the rejection of the true null hypothesis, or a false-positive conclusion. Given that this is a rare disease setting with an a priori 1-sided hypothesis, we will use a 1-sided 5% error rate for the Inhibitor Prevention Trial, assuming that emicizumab is not significantly worse than ELOCTATE. For the Inhibitor Eradication Trial, we have a 2-sided hypothesis, ELOCTATE ITI plus emicizumab differs from ELOCTATE ITI alone, that uses 2.5% of the type 1 error rate on either side. Simulations were used to determine key thresholds to maintain the type 1 error rate and quantify the characteristics of the design needed to maximize the power. To determine thresholds to maintain the type 1 error rate, given the possibility of early success at the interim, simulations mimicked the properties of a standard design (ie, they assumed no prior information to be incorporated into the analysis and 1:1 randomization). After determining those thresholds, design elements of borrowing historical data in the prior and adjusting the allocation ratio were added to maximize the amount of power (see "Power simulations" in supplemental Methods).
For the Inhibitor Prevention Trial, 2 hypotheses will be tested. The primary hypothesis is noninferiority, and the secondary hypothesis is superiority. The design allows the trial to stop early at the interim if there is overwhelming evidence of superiority. Otherwise, the trial proceeds to the final analysis, at which the superiority hypothesis is to be performed sequentially only if the null hypothesis for the noninferiority is rejected. Three decision thresholds need to be defined: (1) at the interim for superiority (after 75% are randomized), (2) at the end of the study for noninferiority, and (3) at the end of the study for superiority. The interim look threshold was set to stop the Inhibitor Prevention Trial when the Bayesian posterior probability that emicizumab is superior to ELOCTATE, based on the data observed in this trial, is ≥99% (Table 2).
INHIBIT Trials Platform: thresholds for maintaining type 1 error
| Study/hypothesis | Posterior probability for superiority to stop at the interim, % | Posterior probability to declare significance at the end of the study, % |
|---|---|---|
| Prevention Trial/noninferiority hypothesis* | >99 | ≥95.7 |
| Prevention Trial/superiority hypothesis† | >99 | ≥95.1 |
| Eradication Trial/superiority hypothesis‡ | <0.005 or >99.5 | <2.3 or >97.7 |
| Study/hypothesis | Posterior probability for superiority to stop at the interim, % | Posterior probability to declare significance at the end of the study, % |
|---|---|---|
| Prevention Trial/noninferiority hypothesis* | >99 | ≥95.7 |
| Prevention Trial/superiority hypothesis† | >99 | ≥95.1 |
| Eradication Trial/superiority hypothesis‡ | <0.005 or >99.5 | <2.3 or >97.7 |
The Prevention Trial noninferiority hypothesis is that emicizumab prophylaxis is noninferior to ELOCTATE prophylaxis in preventing inhibitor development in PUPs with severe hemophilia A over 48 weeks.
The Prevention Trial superiority hypothesis is that emicizumab prophylaxis is superior to ELOCTATE prophylaxis in preventing inhibitor development in PUPs with severe hemophilia A over 48 weeks.
The Eradication Trial superiority hypothesis is that ELOCTATE ITI plus emicizumab is superior to ELOCTATE ITI alone in eradicating inhibitor formation in PTPs with severe hemophilia A and inhibitors over 48 weeks.
If emicizumab is, in fact, 10% worse than ELOCTATE, there is a 0.03% probability of declaring superiority at the interim (a false-positive result for the noninferiority hypothesis). If emicizumab is, in truth, equal to ELOCTATE, there is a 0.15% probability of declaring superiority at the interim (a false positive for the superiority hypothesis). At the end of the trial, these interim error rates must be considered when selecting decision thresholds to control the overall type 1 error rate for the trial.
At the end of the Inhibitor Prevention Trial, the noninferiority test simulations demonstrated that, assuming the trial did not stop at the interim look, the Bayesian posterior probability that emicizumab is noninferior to ELOCTATE needs to be ≥95.7% to keep the error rate at 5%. The superiority test simulations demonstrated that, assuming that the trial did not stop at the interim look, the Bayesian posterior probability that emicizumab is superior to ELOCTATE needs to be ≥95.1% to maintain an error rate of 5%.
For the Inhibitor Eradication Trial, there is 1 2-sided hypothesis: that ELOCTATE ITI plus emicizumab differs from ELOCTATE ITI alone. Using a method similar to that used in the Inhibitor Prevention Trial, the interim stopping rule is to stop when the Bayesian posterior probability that ELOCTATE ITI plus emicizumab is not equal to ELOCTATE ITI alone, given the data to be observed in this trial, is ≥99% (0.5% in the upper tail and 0.5% in the lower tail). For the superiority test we determined that, assuming the trial did not stop at the interim look, the Bayesian posterior probability that ELOCTATE ITI plus emicizumab is superior to ELOCTATE ITI alone needs to be outside of the interval 2.3% to 97.7% (Table 2).
Determining power for the Inhibitor Prevention Trial
With the type 1 error thresholds and the sample size set, 3 factors can be modified to increase power: (1) the amount of borrowing from the prior distribution, (2) the randomization allocation, and (3) the assumptions for the alternative hypothesis. We compared 3 design scenarios after modifying these factors (Figure 4).
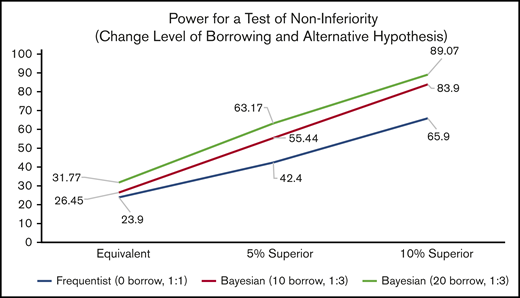
Inhibitor Prevention Trial: power for noninferiority across all design and hypothesis scenarios. The power for a test of noninferiority, assuming various randomization allocation ratios and degrees of “borrowing” for the prior (design scenarios) to determine the power associated with various effect sizes.
Inhibitor Prevention Trial: power for noninferiority across all design and hypothesis scenarios. The power for a test of noninferiority, assuming various randomization allocation ratios and degrees of “borrowing” for the prior (design scenarios) to determine the power associated with various effect sizes.
The first simulated scenario (Design Scenario 1) is the standard frequentist design, which contains no borrowing and 1:1 randomization. This is conceptually the same as a Bayesian analysis with 0 borrowing and 1:1 randomization.
The second scenario (Design Scenario 2) makes 2 changes. First, utilizing the Bayesian design concept of prior distributions, the equivalent of 10 events in 40 subjects is borrowed from the PUPs A-Long trial for the ELOCTATE arm. Second, the ELOCTATE/emicizumab allocation ratio changes from 1:1 to 1:3.
The third scenario (Design Scenario 3) borrows more subjects from the Bayesian prior for the ELOCTATE arm, the equivalent of 20 events in 80 subjects and maintains the ELOCTATE/emicizumab allocation ratio at 1:3.
These 3 design scenarios are calculated under 3 hypothesis scenarios when testing noninferiority: Hypothesis Scenario 1: ELOCTATE and emicizumab have the same effectiveness; Hypothesis Scenario 2: emicizumab is 5% superior to ELOCTATE; and Hypothesis Scenario 3: emicizumab is 10% superior to ELOCTATE.
For the noninferiority test in Table 3, using the standard frequentist framework in Design Scenario 1, the power varies from 24% to 66%, depending on the hypothesis scenario. Although there is an improvement, 66% power is low to plan a definitive trial. Adding the Bayesian priors and changing the allocation ratio to 1:3 (Design Scenario 2), there is only a slight improvement in the scenario in which the 2 arms are equivalent (an increase of 2.5% points), more dramatic improvement when emicizumab is 5% superior (an increase of 13% points), and even more improvement when emicizumab is 10% superior (an increase of 18% points). The power of 83.9% is above the standard 80% power target of standard trials. By increasing the amount of borrowing (from 40 subjects to 80 subjects), there are modest, but steady, increases in power of 5.3%, 7.7%, and 5.2% for the Design Hypothesis 1 (equivalent), Design Hypothesis 2 (5% superior), and Design Hypothesis 3 (10% superior), respectively (Figure 4).
Inhibitor Prevention Trial: power simulations
| Simulation condition | Noninferiority | Superiority | ||||
|---|---|---|---|---|---|---|
| Design scenario | Design 1 | Design 2 | Design 3 | Design 1 | Design 2 | Design 3 |
| No. borrowed | 0 | 40 | 80 | 0 | 40 | 80 |
| Random allocation* | 1:1 | 1:3 | 1:3 | 1:1 | 1:3 | 1:3 |
| Equivalent, % | 23.9 | 26.5 | 31.8 | |||
| 5% superior, % | 42.4 | 55.4 | 63.2 | 11.9 | 9.3 | 11.9 |
| 10% superior, % | 65.9 | 83.9 | 89.1 | 25.4 | 28.8 | 36.2 |
| Simulation condition | Noninferiority | Superiority | ||||
|---|---|---|---|---|---|---|
| Design scenario | Design 1 | Design 2 | Design 3 | Design 1 | Design 2 | Design 3 |
| No. borrowed | 0 | 40 | 80 | 0 | 40 | 80 |
| Random allocation* | 1:1 | 1:3 | 1:3 | 1:1 | 1:3 | 1:3 |
| Equivalent, % | 23.9 | 26.5 | 31.8 | |||
| 5% superior, % | 42.4 | 55.4 | 63.2 | 11.9 | 9.3 | 11.9 |
| 10% superior, % | 65.9 | 83.9 | 89.1 | 25.4 | 28.8 | 36.2 |
With Design Scenario 2, there is between 26.5% and 83.9% power for the noninferiority test, with 66 subjects and α = 0.05. In addition, there is between 9.3% and 28.8% power for the superiority testing.
ELOCTATE/emicizumab.
Determining power for the superiority hypothesis in the Inhibitor Prevention Trial
If the Inhibitor Prevention Trial passes the noninferiority hypothesis (ie, emicizumab is shown to be noninferior to ELOCTATE), the superiority hypothesis will be tested. In the standard frequentist framework in Design Scenario 1, the power varies from 11.9% to 25.4%, depending on the hypothesis scenario. Adding the Bayesian priors (borrowing 40 subjects) and changing the allocation ratio to 1:3 (Design Scenario 2), there is essentially equivalent power when emicizumab is 5% superior (a decrease of 2.6%) and when emicizumab is 10% superior (an increase of 3.4%). Increasing to 80 subjects borrowed (Design Scenario 3), there is essentially equivalent power when emicizumab is 5% superior (an increase of 2.6%) and a small improvement in power when emicizumab is 10% superior (an increase of 7.4%). All of these designs have low power to detect superiority (supplemental Figure 1). The design scenarios have virtually no impact under Hypothesis Scenario 2 (emicizumab is 5% superior) and only a small impact under Hypothesis Scenario 3 (emicizumab is 10% superior).
Determining power for superiority in the Inhibitor Eradication Trial
Because there were no historical data available for the Inhibitor Eradication Trial, we used noninformative priors in both arms, essentially borrowing no data. The randomization allocation ratio is 1:1 to have comparable sample sizes in each group. We assumed that, a priori, the median time to eradicate inhibitors in the ELOCTATE ITI plus emicizumab arms (9.8 weeks) would be about half of the median time to eradicate inhibitors in the ELOCTATE ITI–alone arm (20.6 weeks). This corresponds to event rates of 20% for ELOCTATE ITI and 3.4% event rates for ELOCTATE ITI plus emicizumab. Using the same code as in the Inhibitor Prevention Trial, while incorporating a 2-sided test for difference between arms and the thresholds to maintain the type 1 error from Table 2, the expected power was estimated using simulations (Table 4). Specifically, with 90 subjects, there is >80% power to detect this difference in the median time to eradicate inhibitors.
Inhibitor Eradication Trial: power simulations (n = 90)
| Eradication rate in the emicizumab arm, % | Power, %* |
|---|---|
| 96.0 | 74 |
| 96.3 | 79 |
| 96.6 | 81 |
| 96.9 | 82 |
| Eradication rate in the emicizumab arm, % | Power, %* |
|---|---|
| 96.0 | 74 |
| 96.3 | 79 |
| 96.6 | 81 |
| 96.9 | 82 |
There is 81% power to detect a difference of 96.6% between trial arms, with 90 subjects and α = 0.05.
This assumes the eradication rate in the ELOCTATE ITI arm is 80%.
Discussion
Inhibitor formation is the most serious complication of hemophilia, affecting 25% of those with severe disease, and it is associated with significant morbidity, hospitalizations, and high health care costs. Thus, preventing new inhibitors and eradicating existing inhibitors will promote better health outcomes for children and adults with hemophilia, an approach that, if successful, would be adopted immediately into clinical practice. The drugs that will be compared are approved by the US Food and Drug Administration and are safe and effective in children and adults with hemophilia. Preliminary data from small studies support the need for randomized trials to determine the optimal use of these agents to prevent and eradicate inhibitor formation in severe hemophilia A.
The INHIBIT Clinical Trials Platform is innovative because it uses a Bayesian platform design to compare novel agents in 2 linked randomized phase 3 trials, the Inhibitor Prevention Trial and the Inhibitor Eradication Trial, and it allows for the conduct of these studies in the rare disease hemophilia, for which classical trial design is not practical. The trials incorporate historical data on inhibitor formation, Bayesian priors, which allows for more efficient use of rare subjects and improves power. The 2 trials are integrated for efficiency and conducted in the same centers, with the same staff, visit frequency, blood draws, and laboratory assays. Moreover, as promising new agents emerge, they could potentially be incorporated into the trial platform design. The assays will provide novel mechanistic data on the role of regulatory T cells in promoting and inducing FVIII tolerance and the relationship among FVIII genotype, HLA type, and FVIII levels.
The major disadvantage in the conduct of these trials, despite the advantages noted, is the long timeline to complete trial enrollment and produce definitive results. Despite the use of a platform design, Bayesian priors, and simulations of time-to-event analysis to reduce sample size and incorporate historical data, it is anticipated that it will take up to 6 years to conduct and complete the trials. Further, in the next 6 years there are likely to be other novel subcutaneously administered therapies that are licensed to treat and prevent hemophilia bleeds and avoid FVIII exposure, which might be incorporated into the Bayesian Platform Master Protocol design to compare with the winner of each successive trial to promote better outcomes in preventing and eradicating inhibitors.
Data sharing requests should be sent to Margaret V. Ragni (e-mail: ragni@pitt.edu).
Acknowledgments
The authors thank Anna McGlothlin (Senior Statistical Scientist, Berry Consultants, LLC, Austin TX) for helpful review of the manuscript and Anna McGlothin and Roger Lewis (Senior Statistical Scientist, Berry Consultants, LLC) for expert guidance and many helpful conversations during the design of our clinical trials platform under National Institutes of Health, National Heart, Lung, and Blood Institute grant X01 HL143024-01.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants X01 HL143024-01 (M.V.R.) and U34 HL114674 (M.V.R.).
Authorship
Contribution: M.B. and M.V.R. designed the research, analyzed the findings, and wrote the manuscript; and M.M.B. helped to design the research, analyzed the findings, and edited the final manuscript.
Conflict-of-interest disclosure: M.M.B. is a member of a Data and Safety Monitoring Board for Cerus Corporation. M.V.R. has received research funding from Alnylam Pharmaceuticals, BioMarin, Bioverativ, Sangamo Therapeutics, Spark Therapeutics, and Takeda Pharmaceutical and has served on advisory boards for Alnylam Pharmaceuticals, BioMarin, Bioverativ, and Spark Therapeutics. M.B. declares no competing financial interests.
Correspondence: Margaret V. Ragni, Division of Hematology/Oncology, Department of Medicine and Clinical and Translational Science, University of Pittsburgh, Hemophilia Center of Western PA, 3636 Boulevard of the Allies, Pittsburgh, PA 15213-4306; e-mail: ragni@pitt.edu.

