

Key Points
EZH2 inhibition combined with Bcl-2 inhibition is synergistic in DLBCL subtypes with EZH2 and BCL2 molecular alterations.
Tazemetostat treatment results in upregulation of proapoptotic Bcl-2 family members and priming of mitochondria to apoptotic cell death.

Abstract
Molecular alterations in the histone methyltransferase EZH2 and the antiapoptotic protein Bcl-2 frequently co-occur in diffuse large B-cell lymphoma (DLBCL). Because DLBCL tumors with these characteristics are likely dependent on both oncogenes, dual targeting of EZH2 and Bcl-2 is a rational therapeutic approach. We hypothesized that EZH2 and Bcl-2 inhibition would be synergistic in DLBCL. To test this, we evaluated the EZH2 inhibitor tazemetostat and the Bcl-2 inhibitor venetoclax in DLBCL cells, 3-dimensional lymphoma organoids, and patient-derived xenografts (PDXs). We found that tazemetostat and venetoclax are synergistic in DLBCL cells and 3-dimensional lymphoma organoids that harbor an EZH2 mutation and an IGH/BCL2 translocation but not in wild-type cells. Tazemetostat treatment results in upregulation of proapoptotic Bcl-2 family members and priming of mitochondria to BH3-mediated apoptosis, which may sensitize cells to venetoclax. The combination of tazemetostat and venetoclax was also synergistic in vivo. In DLBCL PDXs, short-course combination therapy resulted in complete remissions that were durable over time and associated with superior overall survival compared with either drug alone.
Introduction
The histone methyltransferase EZH2 has emerged as a therapeutic target in lymphoma.1 Follicular lymphoma (FL) and the germinal center B-cell subtype of diffuse large B-cell lymphoma (DLBCL) express high levels of EZH2 and a subset harbors EZH2 gain-of-function mutations.2-6 Tazemetostat is a small-molecule EZH2 inhibitor that is approved by the US Food and Drug Administration in relapsed/refractory FL.7 Clinical activity in DLBCL is modest,8 suggesting that tazemetostat may need to be combined with other agents for a clinically meaningful antitumor effect.
In DLBCL, EZH2 mutations frequently co-occur with molecular alterations in BCL2, including mutations and/or translocation to the immunoglobulin heavy chain enhancer.9,10 Therefore, targeting both of these oncogenes is a rational therapeutic approach. Venetoclax is a selective Bcl-2 inhibitor that is approved by the US Food and Drug Administration in chronic lymphocytic leukemia and has demonstrated clinical activity in DLBCL.11-13 A previous study using nonselective compounds targeting apoptotic proteins and EZH2 inhibitors suggested that this combination might enhance antilymphoma effects.4 Therefore, we hypothesized that EZH2 and Bcl-2 inhibition would be synergistic in DLBCL.
Methods
Cells and reagents
Tazemetostat and venetoclax were obtained from Selleckchem (Houston, TX). Cell lines, culture conditions, and viability assays are described in supplemental Methods.
Xenografts
SUDHL-6 and patient-derived xenograft (PDX) tumors were implanted subcutaneously in NOD-SCID and NSG mice, respectively. Upon engraftment (tumor volume 200-300 mm3), mice were randomized to vehicle, venetoclax, tazemetostat, or the combination.
Lymphoma organoids
Organoids were generated as previously reported.14-17 Flow cytometry was performed using a LIVE/DEAD Fixable Far Red Dead Cell Stain Kit (Thermo Fisher Scientific; cat. no. L34974). Imaging was performed using calcein-AM and ethidium homodimer. Further details are provided in supplemental Methods.
RNA sequencing analysis and BH3 profiling
Details are provided in supplemental Methods.
Results and discussion
Tazemetostat and venetoclax are synergistic in vitro
To evaluate combination therapy with tazemetostat and venetoclax, we evaluated each drug and the combination in a panel of DLBCL cell lines, including DLBCLs with mutant EZH2 and an IGH/BCL2 translocation (n = 5), wild-type (WT) EZH2 and an IGH/BCL2 translocation (n = 4), and WT EZH2 without a BCL2 translocation (n = 4). All cell lines with an IGH/BCL2 translocation also harbored BCL2 mutations (supplemental Table 1). We observed enhanced cell death after combination therapy compared with either drug alone that was statistically significant in 5 of 6 DLBCLs with alterations in EZH2 and BCL2, 1 of 4 DLBCLs with alterations in BCL2 but not EZH2, and 0 of 5 DLBCLs without alterations in EZH2 or BCL2 (Figure 1A; supplemental Table 2). We next evaluated synergy using the Chou-Talalay method.18 The combination was synergistic in 6 of 6 DLBCLs with EZH2 mutation and BCL2 translocation, 2 of 4 DLBCLs with WT EZH2 and BCL2 translocation, and 0 of 5 DLBCLs without alterations in EZH2 or BCL2 (Figure 1B; supplemental Table 3). This suggests that combination therapy would be most effective in tumors harboring both EZH2 and BCL2 molecular alterations.
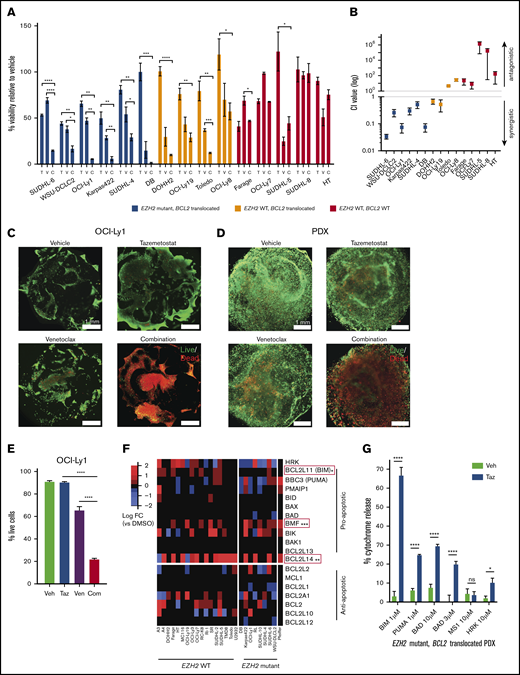
Combination therapy with EZH2 and Bcl-2 inhibitors has in vitro antitumor activity in DLBCL model systems. (A) Cell viability, as measured by CellTiter-Glo, in a panel of DLBCL cell lines treated with vehicle, venetoclax alone (V), tazemetostat alone (T), or venetoclax and tazemetostat in combination (C). Drug dosing for each cell line was optimized to approximate 50% killing (supplemental Table 2). Results represent the mean of 3 biologic replicates, each of which was performed in experimental triplicates. Error bars represent standard error of the mean (SEM). (B) Confidence interval value for DLBCL cells treated with vehicle, venetoclax alone, tazemetostat alone, or venetoclax and tazemetostat in combination over a range of doses at fixed ratios (supplemental Table 3). Cell viability was measured using CellTiter-Glo. Synergy was calculated using CompuSyn. Results represent the mean of 3 biologic replicates, each of which was performed in experimental triplicates. Error bars represent SEM. (C-D) Immunofluorescence of DLBCL organoids treated with vehicle, 50 nM venetoclax, 5 μM tazemetostat, or venetoclax and tazemetostat in combination. Live and dead cells were identified by calcein-AM and ethidium homodimer, respectively, on day 8. Image were obtained on a Nikon TE200U microscope. (E) Percentage of live OCI-Ly1 cells by flow cytometry. Cells were stained for live and dead populations using a LIVE/DEAD Fixable Far Red Dead Cell Stain Kit (Thermo Fisher Scientific; cat. no. L34974). (F) Heat map showing RNA sequencing log2 fold change in BCL2 family members in DLBCL cell lines treated with vehicle vs tazemetostat. The Wilcoxon test was used to determine statistical significance. (G) BH3 profiling of DLBCL PDX cells treated with tazemetostat or vehicle. BIM and PUMA evaluate general priming, BAD evaluates Bcl-2–specific priming, MS1 evaluates MCL-1 priming, and HRK evaluates BCL-XL priming. Error bars represent SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001. Com, combination; ns, not significant; Taz, tazemetostat; Veh, vehicle; Ven, venetoclax.
Combination therapy with EZH2 and Bcl-2 inhibitors has in vitro antitumor activity in DLBCL model systems. (A) Cell viability, as measured by CellTiter-Glo, in a panel of DLBCL cell lines treated with vehicle, venetoclax alone (V), tazemetostat alone (T), or venetoclax and tazemetostat in combination (C). Drug dosing for each cell line was optimized to approximate 50% killing (supplemental Table 2). Results represent the mean of 3 biologic replicates, each of which was performed in experimental triplicates. Error bars represent standard error of the mean (SEM). (B) Confidence interval value for DLBCL cells treated with vehicle, venetoclax alone, tazemetostat alone, or venetoclax and tazemetostat in combination over a range of doses at fixed ratios (supplemental Table 3). Cell viability was measured using CellTiter-Glo. Synergy was calculated using CompuSyn. Results represent the mean of 3 biologic replicates, each of which was performed in experimental triplicates. Error bars represent SEM. (C-D) Immunofluorescence of DLBCL organoids treated with vehicle, 50 nM venetoclax, 5 μM tazemetostat, or venetoclax and tazemetostat in combination. Live and dead cells were identified by calcein-AM and ethidium homodimer, respectively, on day 8. Image were obtained on a Nikon TE200U microscope. (E) Percentage of live OCI-Ly1 cells by flow cytometry. Cells were stained for live and dead populations using a LIVE/DEAD Fixable Far Red Dead Cell Stain Kit (Thermo Fisher Scientific; cat. no. L34974). (F) Heat map showing RNA sequencing log2 fold change in BCL2 family members in DLBCL cell lines treated with vehicle vs tazemetostat. The Wilcoxon test was used to determine statistical significance. (G) BH3 profiling of DLBCL PDX cells treated with tazemetostat or vehicle. BIM and PUMA evaluate general priming, BAD evaluates Bcl-2–specific priming, MS1 evaluates MCL-1 priming, and HRK evaluates BCL-XL priming. Error bars represent SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001. Com, combination; ns, not significant; Taz, tazemetostat; Veh, vehicle; Ven, venetoclax.
Combination therapy yields enhanced activity against 3D DLBCL organoids
Because cell lines in suspension do not reflect the natural architecture of lymphomas, we developed 3-dimensional (3D) lymphoma organoid culture systems composed of extracellular matrix and stromal cells.15,19-21 We studied combination therapy in 2 organoid models: OCI-Ly1 and a primary DLBCL derived from a PDX with an EZH2 mutation and BCL2 translocation22 (supplemental Figure 1). Organoids were treated with tazemetostat, venetoclax, or the combination. In both models, tazemetostat and venetoclax had minimal activity as single agents; however, the combination resulted in significant cell killing, as observed by immunofluorescence using calcein-AM and ethidium homodimer (Figure 1C-D). This was confirmed by flow cytometry in OCI-Ly1 (combo vs venetoclax, P < .0001; combo vs tazemetostat; P < .0001) (Figure 1E).
EZH2 inhibition upregulates proapoptotic Bcl-2 family members and primes mitochondria to apoptosis
To investigate potential mechanisms of synergy, we evaluated previously published RNA sequencing profiles of DLBCL cell lines (n = 26) treated with vehicle or tazemetostat23 to investigate changes in Bcl-2 family members. Tazemetostat-treated cells manifested enhanced expression of the proapoptotic Bcl-2 family members BCL2L11 (P = .012), BMF (P < .001), and BCL2L14 (P = .002) (Figure 1F), suggesting that these may be direct or indirect EZH2 target genes that are derepressed upon EZH2 inhibition. BCL2 inhibition with venetoclax may further enhance proapoptotic signals activated by tazemetostat.
Independent of Bcl-2 family member transcript abundance, sensitivity to Bcl-2 antagonists may also be dependent, in part, on the state of mitochondria in treated cells. Mitochondria primed to outer membrane permeabilization respond more robustly to proapoptotic Bcl-2 family members and have a lower apoptotic threshold.24 We hypothesized that EZH2 inhibition might result in mitochondrial priming. To test this, we performed BH3 profiling in EZH2 mutant BCL2-translocated DLBCL PDX organoids treated with vehicle vs tazemetostat (Figure 1G; supplemental Figure 2A). Tazemetostat-treated cells had increased mitochondrial priming to general apoptotic signaling, as evidenced by cytochrome c release in response to BIM and PUMA (P < .0001). We also observed tazemetostat-induced mitochondrial priming to the Bcl-2–specific peptide BAD (P < .0001). In EZH2 WT PDX organoids, we observed increased priming to BIM, PUMA, and BAD after tazemetostat; however, the change was not as dramatic as in EZH2-mutant tumors, suggesting that EZH2-mutant tumors may be more sensitive to tazemetostat-induced mitochondrial priming (supplemental Figure 2B).
Combined EZH2 and Bcl-2 inhibition eradicates primary human DLBCLs in vivo
To evaluate combination therapy in vivo, we generated SUDHL-6 xenografts and DLBCL22 PDXs, both with mutant EZH2 and a BCL2 translocation. Upon engraftment, mice were randomized to vehicle, either drug alone, or the combination. In SUDHL-6 xenografts, the combination resulted in attenuation of tumor growth compared with either drug alone (combination vs venetoclax, P < .0001; combination vs tazemetostat, P = .0004) (Figure 2A-B). The median overall survival with vehicle, venetoclax, tazemetostat, and the combination was 14, 17, 18, and 42 days, respectively (combination vs venetoclax, P = .0027; combination vs tazemetostat, P = .0018; Figure 2C). Body weight was stable (supplemental Figure 3A).

Inhibition of EZH2 and Bcl-2 in combination results in tumor reduction and prolonged survival in vivo. (A,D,G) Tumor volume over time in SUDHL-6 xenografts and PDXs, as measured by calipers. Error bars represent standard error of the mean (SEM). Tumor growth in SUDHL-6 (B) and PDX (E) xeongrafts, as measured by area under the curve (AUC), for the duration of treatment. Error bars represent SEM. (C,H) Kaplan-Meier curve for overall survival. P values represent comparison with combination therapy for each cohort. Among PDX mice treated with combination therapy, 2 deaths occurred, both in mice without tumors. (F) MRI 3D renderings of PDX mice taken at day 19 and at day 92. Images were obtained using a 1T M3 compact MRI system (Aspect Imaging Ltd.) with a T2-weighted scan without contrast. The tumor area of interest, as determined by automatic thresholding settings, is shown in red. ***P < .001, ****P < .0001.
Inhibition of EZH2 and Bcl-2 in combination results in tumor reduction and prolonged survival in vivo. (A,D,G) Tumor volume over time in SUDHL-6 xenografts and PDXs, as measured by calipers. Error bars represent standard error of the mean (SEM). Tumor growth in SUDHL-6 (B) and PDX (E) xeongrafts, as measured by area under the curve (AUC), for the duration of treatment. Error bars represent SEM. (C,H) Kaplan-Meier curve for overall survival. P values represent comparison with combination therapy for each cohort. Among PDX mice treated with combination therapy, 2 deaths occurred, both in mice without tumors. (F) MRI 3D renderings of PDX mice taken at day 19 and at day 92. Images were obtained using a 1T M3 compact MRI system (Aspect Imaging Ltd.) with a T2-weighted scan without contrast. The tumor area of interest, as determined by automatic thresholding settings, is shown in red. ***P < .001, ****P < .0001.
In PDX mice, we observed complete eradication of tumors, as measured by calipers, in the combination group by day 8 and in the tazemetostat group by day 15. In contrast, we observed progressive tumor growth in venetoclax- or vehicle-treated mice (Figure 2D-E). To determine whether responses were durable after discontinuation of the drug, treatment was discontinued at day 21, and mice were observed. Serial magnetic resonance imaging (MRI) was performed in the tazemetostat and combination cohorts starting at day 19 to determine whether tumors were present that could not be appreciated by calipers. MRI revealed the complete absence of tumor in combination-treated animals but residual tumors in mice treated with tazemetostat alone (Figure 2F). Accordingly, tumors recurred in all mice treated only with tazemetostat (Figure 2F-G; supplemental Figures 4 and 5). Strikingly, after 197 days of follow-up, there was still no detectable disease in combination-treated animals. Median survival in the vehicle, venetoclax, and tazemetostat groups was 43, 43, and 97 days, respectively, but it was not reached in the combination cohort (Figure 2H). Body weight was stable in all cohorts (supplemental Figure 3B).
In summary, using novel model systems, we demonstrate that combined Bcl-2 and EZH2 inhibition results in synergistic antilymphoma effects. We anticipate this combination to be especially effective as precision therapy for the newly identified cluster 3/EZB DLBCL subtype, which frequently harbors EZH2 and BCL2 alterations.9,10 A clinical trial of this combination is currently in development.
Data sharing requests should be sent to Lisa Giulino-Roth (lgr2002@med.cornell.edu).
Acknowledgments
The authors thank the Progressive Assessment of Therapeutics Facility at Cornell University.
This work was supported by National Institutes of Health, National Cancer Institute grant K08 CA219471 (L.G.-R.), The Leukemia & Lymphoma Society Specialized Center of Research Program (L.G.-R., A.M., and E.C.), National Institutes of Health, National Cancer Institute (grant 1R01 CA238745-01A1) (A.S.), The Innovative Molecular Analysis Technology program of the National Institutes of Health, National Cancer Institute (grant 5R33CA212968-03) (A.S.), a National Science Foundation CAREER award (DMR-1554275) (A.S.), and a US Department of Defense Congressionally Directed Medical Research Program Cancer Career Development Award (W81XWH-17-1-0215) (A.S.). R.E.S. was the recipient of a National Science Foundation Graduate Research Fellowship (DGE-1650441).
Authorship
Contribution: H.S., R.E.S., R.R., V.T.-A., S.S., R.P., S.D.B., M.R.T., H.v.B., D.G.-C., S.P., A.L., S.M., A.S., E.C., A.M., and L.G.-R. conducted experiments and/or analyzed data; H.U. provided key reagents; L.G.-R., A.M., E.C., and A.S. designed experiments; H.S. and L.G.-R. wrote the manuscript; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: L.G.-R. has acted as a consultant for Janssen Pharmaceuticals, Celgene, and ADC Therapeutics and has been a member of the scientific advisory board for Merck. A.L. been a member of the scientific advisory board for Zentalis, Flash, and Dialectic; and has a sponsored research agreement with Novartis and AbbVie. A.M. has acted as a consulted for Jubilant, Epizyme, and Constellation, has received research funding from Janssen Pharmaceuticals and Sanofi Aventis, and has acted as an advisor to KDAC Pharma. The remaining authors declare no competing financial interests.
Correspondence: Lisa Giulino-Roth, Weill Cornell Medical College, 525 E 68th St, Payson 695, New York, NY 10065; e-mail: lgr2002@med.cornell.edu.

