

Key Points
Forty-one percent of families with cytopenias and interstitial lung disease and 3% with familial hematologic presentations have a TBD.
Carriers of heterozygous PARN variants often present with lymphocyte telomere lengths at or above the 10th percentile.

Abstract
Telomere biology disorders (TBDs) present heterogeneously, ranging from infantile bone marrow failure associated with very short telomeres to adult-onset interstitial lung disease (ILD) with normal telomere length. Yield of genetic testing and phenotypic spectra for TBDs caused by the expanding list of telomere genes in adults remain understudied. Thus, we screened adults aged ≥18 years with a personal and/or family history clustering hematologic disorders and/or ILD enrolled on The University of Chicago Inherited Hematologic Disorders Registry for causative variants in 13 TBD genes. Sixteen (10%) of 153 probands carried causative variants distributed among TERT (n = 6), TERC (n = 4), PARN (n = 5), or RTEL1 (n = 1), of which 19% were copy number variants. The highest yield (9 of 22 [41%]) was in families with mixed hematologic and ILD presentations, suggesting that ILD in hematology populations and hematologic abnormalities in ILD populations warrant TBD genetic testing. Four (3%) of 117 familial hematologic disorder families without ILD carried TBD variants, making TBD second to only DDX41 in frequency for genetic diagnoses in this population. Phenotypes of 17 carriers with heterozygous PARN variants included 4 (24%) with hematologic abnormalities, 67% with lymphocyte telomere lengths measured by flow cytometry and fluorescence in situ hybridization at or above the 10th percentile, and a high penetrance for ILD. Alternative etiologies for cytopenias and/or ILD such as autoimmune features were noted in multiple TBD families, emphasizing the need to maintain clinical suspicion for a TBD despite the presence of alternative explanations.
Introduction
Since 1999, 14 genes (ACD, CTC1, DKC1, NAF1, NHP2, NOP10, PARN, RTEL1, TERC, TERT, TINF2, STN1, WRAP53, and ZCCHC8) have been identified as the cause of human disorders of defective telomere maintenance.1-8 Inherited defects in each of these genes can cause a range of multiorgan system pathologies now considered part of a single disease entity variably referred to as a telomeropathy, short telomere syndrome, or telomere biology disorder (TBD).4-6 TBD phenotypes of those diagnosed in infancy include severe immunodeficiency, bone marrow failure (BMF), and gastrointestinal and neurologic dysfunction. Those diagnosed in adulthood first present with interstitial lung disease (ILD),9-13 emphysema,14,15 cryptogenic cirrhosis,16,17 isolated macrocytosis or mild cytopenia(s), severe BMF,18,19 and/or hematologic malignancies.18,20,21 Depending on the causative gene, the pattern of inheritance may be X-linked recessive, autosomal dominant, or autosomal recessive with high variability in penetrance and expressivity.4-6
The prevalence of TBD in specific patient populations is actively being defined. For example, the prevalence of the classical TBD presentation, dyskeratosis congenita, featuring the triad of nail dysplasia, skin pigmentation abnormalities, and oral leukoplakia along with peripheral blood lymphocyte telomere lengths less than the first percentile for age, is rare.22 In comparison, 1% of adults with emphysema,15 3% to 10% of adults with ILD,9,13 and up to 30% of those with familial pulmonary fibrosis,14 defined as 2 or more cases of ILD in first- or second-degree relatives, harbor a pathogenic (P) or likely pathogenic (LP) variant in a known TBD gene.
Prevalence in adults with hematologic disorders is less well characterized. Most studies have examined TERT and TERC only, and lack of access to germline tissue has limited broad genetic testing of patients with active hematologic malignancies. With these caveats, ∼1% to 5% of those with aplastic anemia (AA)18,19,23 and 1% to 3% of those with myelodysplastic syndrome (MDS)/acute leukemia carry an inherited P/LP TBD gene variant.18,21,23 The single series published to date in families clustering AA/MDS/acute leukemia found P/LP variants in TERT or TERC in 5 (19%) of 27, suggesting that this is a high-yield testing scenario.24 Given that patients with TBD are prone to serious complications of standard treatments for sporadic AA/MDS/acute leukemia and ILD,25-28 knowing whom to test as well as having sensitive and specific diagnostic tests to identify these patients are critical.
When present within an individual or in close relatives in a family, cooccurrence of hematologic and pulmonary symptoms has been shown to increase the likelihood of identifying a causative variant in TERT or TERC. In one series, when both overt AA and ILD were present, all 10 families had an identifiable TERT or TERC variant.29 Diaz de Leon et al30 showed that asymptomatic TERT variant carriers often have combined subclinical pulmonary and hematologic features, including reduced diffusion capacity as well as mild thrombocytopenia and larger red blood cell mean corpuscular volume than their wild-type relatives. If hematologic and pulmonary manifestations also frequently cooccur in those carrying P/LP variants in TBD genes beyond TERT and TERC, this personal and/or family history combination may be especially useful in selecting whom to test.
From a clinical diagnostic perspective, critically short telomeres (less than the first percentile for age) in peripheral blood lymphocytes measured by flow cytometry and fluorescence in situ hybridization (flowFISH)31 can be highly sensitive and specific for a TBD diagnosis.4,32 However, telomere length interpretation in those with active hematologic malignancies is complicated by the effect of the hematologic malignancy itself on telomere length. Furthermore, adults with genetically diagnosed TBD presenting with ILD, mild blood count abnormalities, or MDS/acute leukemia often have lymphocyte telomere lengths between the first and 10th percentile and occasionally well above the 10th percentile.22,33,34 Because these values overlap the lower end of the normal reference range from healthy populations, it is frequently challenging to know which patients are likely to have a TBD and which are not.
Challenges of telomere length interpretation, the lack of classical mucocutaneous features in most, and the similarity between typical age-related and TBD-related comorbidities make it difficult to provide a clinical TBD diagnosis without genetic testing and detailed family assessment in adult populations. Prevalence of TBD and phenotype among adults with hematologic diagnoses are particularly understudied. Thus, we sought to determine the prevalence and phenotype of TBD in adults with hematologic presentations that should be enriched for TBD, those with familial clustering of chronic hematologic abnormalities, and/or AA/MDS/acute leukemia with or without ILD through comprehensive genomic screening of 13 TBD genes and family assessments. We describe phenotype and lymphocyte telomere lengths measured by using flowFISH in 16 TBD families, including 5 carrying heterozygous PARN variants.
Methods
Patient population
Probands aged ≥18 years who did not have a known genetic diagnosis at the time of enrollment on The University of Chicago Inherited Hematologic Disorders Registry between 2011 and August 2019 were eligible. Those with a personal history of chronic unexplained cytopenia(s) or macrocytosis, AA/MDS/acute leukemia, and/or ILD fitting one of the following definitions were included: (1) familial hematologic disorder cases, defined as 2 or more cases of chronic cytopenia/AA/MDS/acute leukemia in first- and/or second-degree relatives or a proband with AA/MDS/acute leukemia with at least one additional nonpulmonary TBD feature; (2) hematologic disorder and ILD overlap cases, defined as at least one case of cytopenia/AA/MDS/acute leukemia and at least one case of ILD in a proband or in first- and/or second-degree relatives; and (3) familial pulmonary fibrosis cases, defined as 2 or more cases of ILD without hematologic manifestations in first- and/or second-degree relatives (supplemental Table 1). Informative close relatives were enrolled whenever possible. Medical, exposure, and family history were obtained by patient interview and review of medical records. Imaging studies (by M.E.S. and A.A.), lung pathology (by A.N.H.), and hematopathology (by S. Gurbuxani) were obtained and centrally reviewed. Informed consent was obtained from all participants. This study was approved by The University of Chicago Institutional Review Board.
Genetic testing
Patient DNA extracted from cultured skin fibroblasts, peripheral blood, or saliva underwent sequencing and analysis of ACD, CTC1, DKC1, NAF1, NHP2, NOP10, PARN, RTEL1, TERC, TERT, TINF2, STN1, and WRAP53 as part of larger hereditary hematologic malignancy/BMF panels either by targeted genomic capture and gene panel–based or whole-exome sequencing at The University of Chicago or at The University of Washington (supplemental Table 1).35,36 Due to updating of genetic panels over time as new genes were discovered, ACD and/or PARN were not covered in 28 patients. Copy number variants were assessed by either next-generation sequencing or custom high-density gene-targeted array comparative genomic hybridization. Sanger sequencing, multiplex ligation-dependent probe amplification, or quantitative real-time polymerase chain reaction (PCR) were used for validation of P/LP single nucleotide variants, insertions/deletions, or copy number variants, as well as for segregation analysis within a family. Variants were classified by using The American College of Medical Genetics and Genomics/The Association for Molecular Pathology guidelines and the Clinical Genome Sequence Variant Interpretation Working Group recommendations.37-42 Criteria were tailored in a gene-specific manner as detailed in the supplemental Methods and supplemental Tables 2 and 3.
Telomere length measurements
All peripheral blood lymphocyte telomere lengths were measured by using flowFISH in the Johns Hopkins Pathology Laboratories, except for proband 875 III.1, whose telomeres were measured at Repeat Diagnostics Inc. using methods previously described.22,31
Quantitative real-time PCR
Primers were used to amplify patient DNA for specific exons of BFAR, PARN, PLA2G10, and NOMO1 using the StepOnePlus Real-Time PCR System (Applied Biosystems) in triplicate. PCR products were analyzed with StepOne Software v2.3 (Applied Biosystems). One no template control was included per primer pair per run. Amplification efficiency was calculated for each run by using serial dilutions. The standard 2-ΔΔCT method was used to calculate copy number ratio.
Statistics
Two-sided Fisher’s exact tests were used to test the difference between categorical variables using Stata version 16 (StataCorp).
Results
Prevalence of P/LP variants vary by phenotype
In total, 153 adult probands met eligibility criteria (Figure 1; supplemental Table 1). Of these, 139 included a personal or family history of at least 1 hematologic manifestation; 117 met the familial hematologic disorder definition, and 22 met the mixed hematologic disorder and ILD overlap case definition. The remaining 14 fit familial pulmonary fibrosis criteria. The median age at initial hematologic and/or ILD diagnosis among all families was 50.8 years (range, 4-82 years) and 60.7 years (range, 41-78 years), respectively.
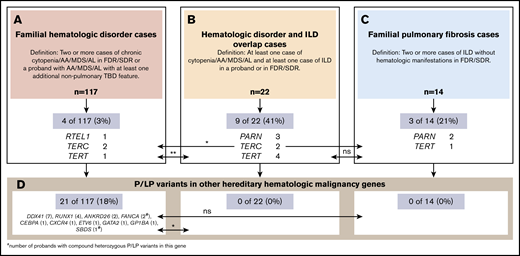
Comparison of the prevalence of causative TBD and other gene variants by familial hematologic and/or ILD presentations in adults. A total of 153 probands were tested and are grouped according to their phenotypic presentation. (A) Familial hematologic disorder cases. (B) Mixed hematologic disorder and ILD cases. (C) Familial pulmonary fibrosis cases. The proportions of probands found to carry P or LP variants in telomere genes (A-C) or other hereditary hematologic malignancy syndrome genes (D) by clinical presentation overall and by specific gene are provided and compared by using 2-sided Fisher’s exact tests. Levels of significance are shown as: *P < .05, **P < .001. AL, acute leukemia; FDR, first-degree relative; ns, not significant; SDR, second-degree relative.
Comparison of the prevalence of causative TBD and other gene variants by familial hematologic and/or ILD presentations in adults. A total of 153 probands were tested and are grouped according to their phenotypic presentation. (A) Familial hematologic disorder cases. (B) Mixed hematologic disorder and ILD cases. (C) Familial pulmonary fibrosis cases. The proportions of probands found to carry P or LP variants in telomere genes (A-C) or other hereditary hematologic malignancy syndrome genes (D) by clinical presentation overall and by specific gene are provided and compared by using 2-sided Fisher’s exact tests. Levels of significance are shown as: *P < .05, **P < .001. AL, acute leukemia; FDR, first-degree relative; ns, not significant; SDR, second-degree relative.
Overall, 16 (10%) probands were found to carry a P (n = 5) or LP (n = 11) heterozygous variant in a TBD gene (Figure 1; Table 1; supplemental Table 3), distributed among TERT (n = 6), TERC (n = 4), PARN (n = 5), and RTEL1 (n = 1). Nine (56%) of the 16 variants were novel. All except for 1 variant (TERC n.114_115del, which was identified in 2 seemingly unrelated families [218 and 684]) were unique to a single family. Three (19%) of the 16 variants were copy number variants, including a whole-gene deletion of TERC (supplemental Figure 1), a large deletion encompassing exons 1-21 of PARN as well as several genes upstream (Figure 2A-B; supplemental Figure 2), and a deletion of exon 12 of PARN. Single TBD gene variants of uncertain significance were identified in 9 (6%) probands, one of whom (875) also carried an LP TERC variant, distributed among CTC1 (3), NAF1 (1), PARN (1), RTEL1 (2), TERT (1), and WRAP53 (1).
Hematologic, pulmonary, and other characteristics of confirmed carriers of P or LP telomere gene variants in 16 families
| Pedigree ID | Proband/relative | Sex | Gene | Variant cDNA | Variant protein | Variant class | Hematologic phenotype | Age at diagnosis of hematologic disease, y | Pulmonary phenotype | Age at diagnosis of pulmonary disease, y | Comorbidities | Autoimmune features | PB lymphocyte telomere length (flowFISH, percentile) | Variant previously reported (PMID if yes) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 557 II.1 | Proband | M | PARN | c.24del | p.F8Lfs*12 | P | Polycythemia | 52 | ILD/UIP | 51 | AbTPO↑ | <1st | 29463756 | |
| 1080 III.1 | Proband | M | PARN | c.709C>T | p.R237* | P | Macrocytosis | 47 | ILD/UIP | 52 | GERD | 10th | 26810774, 28099038 | |
| 1080 II.1 | Aunt | F | Macrocytosis | ILD | 77 | 10th-50th | ||||||||
| 1080 II.2 | Aunt | F | ILD | 74 | 10th-50th | |||||||||
| 1080 II.3 | Aunt | F | ILD | 62 | GERD | 10th-50th | ||||||||
| 1080 II.4 | Aunt | F | 1st-10th | |||||||||||
| 841 II.1 | Proband | M | PARN | c.1006-2A>G | p.? | LP | BM dyserythropoiesis | 54 | ILD/UIP | 49 | 10th | No | ||
| 832 III.1 | Proband | M | PARN | Deletion exon 1-21 | NA | P | ILD/UIP and constrictive bronchiolitis | 61 | 50th | No | ||||
| 832 III.3 | Brother | M | ILD/UIP | 67 | ND | |||||||||
| 832 III.4 | Brother | M | ILD | 63 | 1st-10th | |||||||||
| 832 III.7 | Sister | F | ND | |||||||||||
| 832 III.9 | Cousin | F | ND | |||||||||||
| 832 III.11 | Cousin | F | ND | |||||||||||
| 832 III.12 | Cousin | F | ND | |||||||||||
| 832 III.13 | Cousin | F | ILD/UIP | 63 | Osteoporosis, elevated LFT results | 10th-50th | ||||||||
| 832 IV.6 | Niece | F | 10th-50th | |||||||||||
| 1210 II.1 | Proband | M | PARN | Deletion exon 12 | NA | LP | Intermittent thrombocytopenia | 56 | ILD (unclassifiable) | 56 | GERD, small hiatal hernia, generalized anxiety disorder/ depression upon ILD diagnosis | ANA 1:320 (speckled and nucleolar), ACE↑, CCP↑ | <1st | No |
| 909 II.1 | Proband | M | RTEL1 | c.1135+1G>A | p.? | P | Gastric DLBCL, AML | 48 | early graying | <1st | No | |||
| 684 II.1 | Proband | M | TERC | n.114_115del | NA | LP | Chronic pancytopenia/AA with macrocytosis | 18 | ND | 27192671 | ||||
| 218 III.1 | Proband | M | TERC | n.114_115del | NA | LP | Neutropenia | Mildly reduced diffusion capacity | 31 | 1st | 27192671 | |||
| 218 II.1 | Mother | F | Chronic pancytopenia/AA | 40 | ILD | 60 | ND | |||||||
| 875 III.1 | Proband | M | TERC | n.357_365del | NA | LP | Pancytopenia, MDS (MDS-U) | 40, 53 | 20th | No | ||||
| 775 III.1 | Proband | M | TERC | Whole-gene deletion | NA | LP | Chronic neutropenia, macrocytosis, intermittent thrombocytopenia, hypocellular bone marrow, CD3/CD4 lymphocytopenia, low IgM | 14 | Pancreatic insufficiency (sweat chloride normal), fine reticular skin hypopigmentation | ANA 1:320 (speckled) | <1st | No | ||
| 865 II.1 | Proband | F | TERT | c.230T>C | p.L77P | LP | ILD/CHP | 73 | SCC (skin), skin hypopigmentation, osteoporosis | ILD/CHP, psoriasis | <1st | No | ||
| 865 II.2 | Brother | M | ILD/CHP | 74 | ILD/CHP | ND | ||||||||
| 865 II.3 | Sister | F | Asthma | ND | ||||||||||
| 865 III.1 | Son | M | ILD/CHP | 53 | ILD/CHP | ND | ||||||||
| 913 II.4 | Proband | M | TERT | c.347C>T | p.T116I | LP | ILD/IPAF | 55 | SCC tongue, osteoporosis with multiple compression fractures, GERD | ILD/IPAF, ANA 1:320 (speckled), Raynaud syndrome | <1st | No | ||
| 913 III.5 | Daughter | F | Raynaud syndrome | <1st | ||||||||||
| 913 III.4 | Daughter | F | <1st | |||||||||||
| 913 II.2 | Sister | F | Osteoporosis | ND | ||||||||||
| 913 II.1 | Sister | F | Macrocytosis | 61 | Osteoporosis | 1st-10th | ||||||||
| 913 III.1 | Nephew | M | asthma | <1st | ||||||||||
| 913 III.3 | Niece | F | <1st | |||||||||||
| 246 II.1 | Proband | M | TERT | c.1891C>T | p.R631W | P | AML with MDS features | 75 | ND | 18460650, 26859482 | ||||
| 088 II.4 | Proband | F | TERT | c.1990G>C | p.V664L | LP | MDS (complex karyotype) | 56 | ND | 29463756 | ||||
| 088 II.1 | Brother | M | Pancytopenia with macrocytosis | 1st-10th | ||||||||||
| 088 II.2 | Sister | F | MDS (RARS) | 62 | Skin hypopigmentation, osteoporosis | Pernicious anemia | 1st-10th | |||||||
| 088 III.2 | Niece | F | <1st | |||||||||||
| 669 III.1 | Proband | M | TERT | c.2011C>G | p.R671G | LP | Macrocytic anemia, thrombocytopenia (CCUS with acquired U2AF1 variant) | ILD/UIP | 51 | ANA 1:160 (hom/speckled), Aldolase↑ | <1st | No | ||
| 669 III.2 | Brother | M | Thrombocytopenia | 48 | Liver fibrosis | Near 1st | ||||||||
| 1020 II.1 | Proband | F | TERT | c.2812C>T | p.R938W | LP | Macrocytic anemia | ILD/UIP | 48 | Early graying | <1st | 28099038 |
| Pedigree ID | Proband/relative | Sex | Gene | Variant cDNA | Variant protein | Variant class | Hematologic phenotype | Age at diagnosis of hematologic disease, y | Pulmonary phenotype | Age at diagnosis of pulmonary disease, y | Comorbidities | Autoimmune features | PB lymphocyte telomere length (flowFISH, percentile) | Variant previously reported (PMID if yes) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 557 II.1 | Proband | M | PARN | c.24del | p.F8Lfs*12 | P | Polycythemia | 52 | ILD/UIP | 51 | AbTPO↑ | <1st | 29463756 | |
| 1080 III.1 | Proband | M | PARN | c.709C>T | p.R237* | P | Macrocytosis | 47 | ILD/UIP | 52 | GERD | 10th | 26810774, 28099038 | |
| 1080 II.1 | Aunt | F | Macrocytosis | ILD | 77 | 10th-50th | ||||||||
| 1080 II.2 | Aunt | F | ILD | 74 | 10th-50th | |||||||||
| 1080 II.3 | Aunt | F | ILD | 62 | GERD | 10th-50th | ||||||||
| 1080 II.4 | Aunt | F | 1st-10th | |||||||||||
| 841 II.1 | Proband | M | PARN | c.1006-2A>G | p.? | LP | BM dyserythropoiesis | 54 | ILD/UIP | 49 | 10th | No | ||
| 832 III.1 | Proband | M | PARN | Deletion exon 1-21 | NA | P | ILD/UIP and constrictive bronchiolitis | 61 | 50th | No | ||||
| 832 III.3 | Brother | M | ILD/UIP | 67 | ND | |||||||||
| 832 III.4 | Brother | M | ILD | 63 | 1st-10th | |||||||||
| 832 III.7 | Sister | F | ND | |||||||||||
| 832 III.9 | Cousin | F | ND | |||||||||||
| 832 III.11 | Cousin | F | ND | |||||||||||
| 832 III.12 | Cousin | F | ND | |||||||||||
| 832 III.13 | Cousin | F | ILD/UIP | 63 | Osteoporosis, elevated LFT results | 10th-50th | ||||||||
| 832 IV.6 | Niece | F | 10th-50th | |||||||||||
| 1210 II.1 | Proband | M | PARN | Deletion exon 12 | NA | LP | Intermittent thrombocytopenia | 56 | ILD (unclassifiable) | 56 | GERD, small hiatal hernia, generalized anxiety disorder/ depression upon ILD diagnosis | ANA 1:320 (speckled and nucleolar), ACE↑, CCP↑ | <1st | No |
| 909 II.1 | Proband | M | RTEL1 | c.1135+1G>A | p.? | P | Gastric DLBCL, AML | 48 | early graying | <1st | No | |||
| 684 II.1 | Proband | M | TERC | n.114_115del | NA | LP | Chronic pancytopenia/AA with macrocytosis | 18 | ND | 27192671 | ||||
| 218 III.1 | Proband | M | TERC | n.114_115del | NA | LP | Neutropenia | Mildly reduced diffusion capacity | 31 | 1st | 27192671 | |||
| 218 II.1 | Mother | F | Chronic pancytopenia/AA | 40 | ILD | 60 | ND | |||||||
| 875 III.1 | Proband | M | TERC | n.357_365del | NA | LP | Pancytopenia, MDS (MDS-U) | 40, 53 | 20th | No | ||||
| 775 III.1 | Proband | M | TERC | Whole-gene deletion | NA | LP | Chronic neutropenia, macrocytosis, intermittent thrombocytopenia, hypocellular bone marrow, CD3/CD4 lymphocytopenia, low IgM | 14 | Pancreatic insufficiency (sweat chloride normal), fine reticular skin hypopigmentation | ANA 1:320 (speckled) | <1st | No | ||
| 865 II.1 | Proband | F | TERT | c.230T>C | p.L77P | LP | ILD/CHP | 73 | SCC (skin), skin hypopigmentation, osteoporosis | ILD/CHP, psoriasis | <1st | No | ||
| 865 II.2 | Brother | M | ILD/CHP | 74 | ILD/CHP | ND | ||||||||
| 865 II.3 | Sister | F | Asthma | ND | ||||||||||
| 865 III.1 | Son | M | ILD/CHP | 53 | ILD/CHP | ND | ||||||||
| 913 II.4 | Proband | M | TERT | c.347C>T | p.T116I | LP | ILD/IPAF | 55 | SCC tongue, osteoporosis with multiple compression fractures, GERD | ILD/IPAF, ANA 1:320 (speckled), Raynaud syndrome | <1st | No | ||
| 913 III.5 | Daughter | F | Raynaud syndrome | <1st | ||||||||||
| 913 III.4 | Daughter | F | <1st | |||||||||||
| 913 II.2 | Sister | F | Osteoporosis | ND | ||||||||||
| 913 II.1 | Sister | F | Macrocytosis | 61 | Osteoporosis | 1st-10th | ||||||||
| 913 III.1 | Nephew | M | asthma | <1st | ||||||||||
| 913 III.3 | Niece | F | <1st | |||||||||||
| 246 II.1 | Proband | M | TERT | c.1891C>T | p.R631W | P | AML with MDS features | 75 | ND | 18460650, 26859482 | ||||
| 088 II.4 | Proband | F | TERT | c.1990G>C | p.V664L | LP | MDS (complex karyotype) | 56 | ND | 29463756 | ||||
| 088 II.1 | Brother | M | Pancytopenia with macrocytosis | 1st-10th | ||||||||||
| 088 II.2 | Sister | F | MDS (RARS) | 62 | Skin hypopigmentation, osteoporosis | Pernicious anemia | 1st-10th | |||||||
| 088 III.2 | Niece | F | <1st | |||||||||||
| 669 III.1 | Proband | M | TERT | c.2011C>G | p.R671G | LP | Macrocytic anemia, thrombocytopenia (CCUS with acquired U2AF1 variant) | ILD/UIP | 51 | ANA 1:160 (hom/speckled), Aldolase↑ | <1st | No | ||
| 669 III.2 | Brother | M | Thrombocytopenia | 48 | Liver fibrosis | Near 1st | ||||||||
| 1020 II.1 | Proband | F | TERT | c.2812C>T | p.R938W | LP | Macrocytic anemia | ILD/UIP | 48 | Early graying | <1st | 28099038 |
All variants have been detected in a heterozygous state and were described according to the recommendations of the Human Genome Variation Society using the following transcripts: TERC NR_001566.1, TERT NM_198253.2, RTEL1 NM_001283009.1, and PARN NM_002582.3. Confirmed variant carriers from each individual family are organized in a single gray or white band with row names indicating individual and relationship.
ACE, angiotensin-converting enzyme; ANA, antinuclear antibody; BM, bone marrow; CCP, cyclic citrullinated peptide; CCUS, clonal cytopenia of undetermined significance; cDNA, complementary DNA; CHP, chronic hypersensitivity pneumonitis; DLBCL, diffuse large B-cell lymphoma; GERD, gastroesophageal reflux disease; hom, homogenous; IgM, immunoglobulin M; IPAF, interstitial pneumonia with autoimmune features; LFT, liver function test; MDS-U, MDS, unclassifiable; NA, not applicable; ND, not done; PB, peripheral blood; PMID, PubMed identifier; RARS, refractory anemia with ring sideroblasts; SCC, squamous cell carcinoma; UIP, usual interstitial pneumonia.
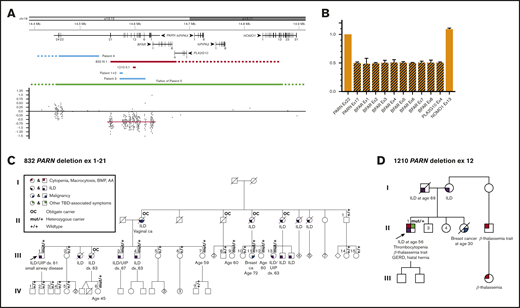
Large heterozygous PARN deletions can be tolerated without neuropsychiatric disorders but may feature cytopenias. (A) Schematic of the genomic region surrounding the deletion and location and distribution of the microarray probes. The genomic region displayed here ranges from 14.4 to 14.9 Mb on the short arm of chromosome 16. All 6 protein-coding genes and their distribution of exons are shown in the upper part of the schematic. The middle part includes the deletions of proband 832 III.1 and proband 1210 II.1 found in our study (red bars) determined by microarray and/or quantitative real-time PCR. The distal breakpoint of the deletion ex1-21 is located upstream of PARN within the dashed bar between exon 4 of PLA2G10 and exon 13 of NOMO1. The blue bars indicate the previously described 4 intragenic deletions of PARN in the study from Dhanraj et al45 with the proximal breakpoint in patient 4 located further downstream (dashed bar). The green bar represents the previously reported whole-gene deletion in the study from Moon et al46 with both the proximal and distal breakpoints located outside of the gene schematic (dashed bar). The bottom part displays the distribution and location of the microarray probes used for proband 832 III.1. The mean log2 ratio of the deleted probes is −0.65, and the deleted region is highlighted with a red bar. (B) Mapping of the distal breakpoint. Quantitative real-time PCR results of the relative copy number of exon 17 of PARN, exon 1-8 of BFAR, exon 4 of PLA2G10, and exon 13 of NOMO1 compared with the non-deleted exon 22 of PARN. Values at or around 0.5 indicate a heterozygous loss, and values at or around 1 indicate the presence of both alleles. (C-D) Pedigrees of a large family with a heterozygous deletion of exons 1-21 of the PARN gene and a family with a single exon PARN deletion. A circle represents a woman, and a square represents a man. Roman numerals indicate generations (eg, I, II, III, IV). A slash through the circle or square indicates that the person is deceased. The proband is indicated by the arrow. The variant carrier status is mut/+ for carriers and +/+ for wild type. OC indicates an individual who is an obligate carrier of the familial PARN deletion. The phenotype is detailed below the symbol. The pedigree number and variant found in each family are noted above the pedigree.
Large heterozygous PARN deletions can be tolerated without neuropsychiatric disorders but may feature cytopenias. (A) Schematic of the genomic region surrounding the deletion and location and distribution of the microarray probes. The genomic region displayed here ranges from 14.4 to 14.9 Mb on the short arm of chromosome 16. All 6 protein-coding genes and their distribution of exons are shown in the upper part of the schematic. The middle part includes the deletions of proband 832 III.1 and proband 1210 II.1 found in our study (red bars) determined by microarray and/or quantitative real-time PCR. The distal breakpoint of the deletion ex1-21 is located upstream of PARN within the dashed bar between exon 4 of PLA2G10 and exon 13 of NOMO1. The blue bars indicate the previously described 4 intragenic deletions of PARN in the study from Dhanraj et al45 with the proximal breakpoint in patient 4 located further downstream (dashed bar). The green bar represents the previously reported whole-gene deletion in the study from Moon et al46 with both the proximal and distal breakpoints located outside of the gene schematic (dashed bar). The bottom part displays the distribution and location of the microarray probes used for proband 832 III.1. The mean log2 ratio of the deleted probes is −0.65, and the deleted region is highlighted with a red bar. (B) Mapping of the distal breakpoint. Quantitative real-time PCR results of the relative copy number of exon 17 of PARN, exon 1-8 of BFAR, exon 4 of PLA2G10, and exon 13 of NOMO1 compared with the non-deleted exon 22 of PARN. Values at or around 0.5 indicate a heterozygous loss, and values at or around 1 indicate the presence of both alleles. (C-D) Pedigrees of a large family with a heterozygous deletion of exons 1-21 of the PARN gene and a family with a single exon PARN deletion. A circle represents a woman, and a square represents a man. Roman numerals indicate generations (eg, I, II, III, IV). A slash through the circle or square indicates that the person is deceased. The proband is indicated by the arrow. The variant carrier status is mut/+ for carriers and +/+ for wild type. OC indicates an individual who is an obligate carrier of the familial PARN deletion. The phenotype is detailed below the symbol. The pedigree number and variant found in each family are noted above the pedigree.
Prevalence of an inherited P/LP variant in a TBD gene differed according to phenotype. Among all 139 families with at least 1 hematologic manifestation (Figure 1A-B), 13 (10%) carried a P/LP variant. A significantly greater proportion were found in families in which hematologic disorders and ILD overlap compared with those with familial hematologic disorder presentations without ILD (9 of 22 [41%] vs 4 of 117 [3%], respectively; P < .01). The proportion in overlap cases was similar to that in familial pulmonary fibrosis families without hematologic manifestations (9 of 22 [41%] vs 3 of 14 [21%], respectively; P = .29) (Figure 1B-C).
In contrast, 21 (18%) of 117 families with familial hematologic disorders without ILD carried P/LP variants in other hereditary hematologic malignancy syndrome genes vs none with overlap or familial pulmonary fibrosis presentations (P = .03 and P = .12, respectively) (Figure 1D; supplemental Table 1). The 21 variants were distributed among DDX41 (n = 7 [6%]), RUNX1 (n = 4 [3%]), ANKRD26 (n = 2 [2%]), FANCA (n = 2 compound heterozygous [2%]), and 1 (1%) each in CEBPA, CXCR4, ETV6, GATA2, GP1BA (heterozygous), and SBDS (compound heterozygous) (Figure 1). Although the overall prevalence of P/LP TBD gene variants in familial hematologic disorders without ILD is modest (n = 4 [3%]), cumulatively, this proportion ranks second behind DDX41 and is tied with RUNX1 as the most frequently identified disorders in this setting.
Hematologic presentations among adult carriers of P/LP variants
Among the 13 families with inherited P/LP TBD gene variants with overlap or familial hematologic disorder presentations, 5 (31%) families reported at least 1 hematologic malignancy, of which 4 clustered multiple cases of MDS and/or acute leukemias (Table 1; supplemental Table 4; supplemental Figure 3A-E). The remaining 9 had single or multiple individuals with macrocytosis/macrocytic anemia, thrombocytopenia, neutropenia, and/or multiple cytopenias.
Blood counts were available for 24 of 42 confirmed P/LP TBD variant carriers without active hematologic malignancy (supplemental Table 4). The complete blood count with differential was completely normal in 11 (46%) carriers. The most common abnormalities found were: macrocytosis (n = 7 [29%]), thrombocytopenia (n = 5 [21%]), neutropenia (n = 5 [21%]), and anemia (n = 3 [13%]). Neutropenia was common among TERC P/LP variant carriers, observed in all 4 carriers without hematologic malignancy, whereas neutropenia was noted in only 1 of 9 and 0 of 10 TERT and PARN variant carriers, respectively. Other identifiable possible contributors to observed cytopenias were present in 3 cases and included: mild splenomegaly in 1 patient with thrombocytopenia (669 III.2), an acquired U2AF1 variant in 1 patient with macrocytic anemia and thrombocytopenia (669 III.1) (Table 1), and β-thalassemia trait (1210 II.1) as an explanation for the patient’s mild microcytic anemia but not for the intermittent thrombocytopenia.
Hematologic malignancies were confirmed in 5 carriers. The median age of onset of hematologic malignancy was 58.8 years (range, 48-75 years). Specific diagnoses included: 1 patient (909 II.1) who carried a P heterozygous RTEL1 variant with concurrent diagnoses of acute myeloid leukemia (AML) and gastric diffuse large B-cell lymphoma, 3 patients with P/LP TERT variants with MDS or AML (1 each with MDS with complex karyotype, MDS-RARS, and AML with myelodysplasia-related changes), and 1 patient with an LP TERC variant with MDS-unclassifiable (supplemental Tables 1 and 4).
Hematologic, pulmonary, and other manifestations in carriers of heterozygous P/LP PARN variants
Among the 17 heterozygous P/LP PARN variant carriers identified in this study, 2 had macrocytosis, 1 had transient thrombocytopenia, and 1 had polycythemia as baseline blood count abnormalities (Table 1; supplemental Table 4; Figures 2C-D,and 3A-C). No hematologic malignancies were reported. Bone marrow aspirate performed before lung transplantation on proband 841 II.1 revealed dyserythropoiesis with megaloblastoid maturation (supplemental Figure 4). He reported a family history of childhood-onset BMF requiring transplantation in a distant relative, but carrier status of this individual could not be confirmed. Other TBD organ system manifestations were minimal, with 1 of 17 (832 III.13) reporting osteoporosis and elevated liver function test results. Two family members whose genotype was unknown had had intellectual disability along with ILD or death in infancy due to liver and lung abnormalities.
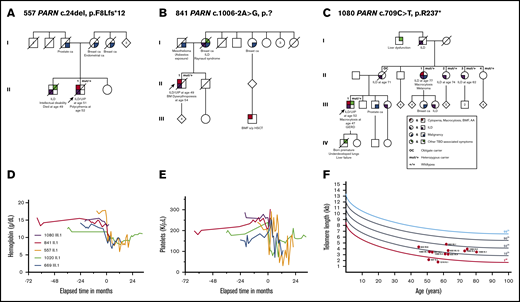
Hematologic abnormalities and telomere lengths in carriers of heterozygous variants in PARN. (A-C) Pedigrees of families with segregating PARN variants. A circle represents a woman, and a square represents a man. Roman numerals indicate generations (I, II, III, IV). A slash through the circle or square indicates the person is deceased. The proband is indicated by the arrow. The variant carrier status is mut/+ for carriers and +/+ for wild type. OC indicates an individual who is an obligate carrier of the familial PARN variant. Color-shaded quarters represent different phenotypes differentiated by color: red stands for cytopenia, AA, or BMF; purple stands for ILD; blue indicates the diagnosis of a malignancy; and green marks all other TBD-associated symptoms. The exact disease/phenotype is detailed below the symbol. Pedigree numbers and the variant found in each family are all designated above the pedigree. Longitudinal hemoglobin (D) and platelet count (E) in patients pre– and post–lung transplantation. Spaghetti plots depict longitudinal blood counts from 3 PARN variant carriers (purple, red, orange) and 2 TERT variant carriers (green, blue) pre– and post–lung transplantation. The time of double-lung transplant is indicated as time point 0, and negative and positive values on the x-axis indicate the number of months before or after transplantation. (F) Peripheral blood lymphocyte telomere lengths as measured by flowFISH. Telomere length from each individual variant carrier is displayed as a red dot with his or her respective pedigree and individual ID number. Each individual’s age-adjusted telomere length is plotted compared with the testing laboratory’s population of healthy age-adjusted control subjects (in percentiles). HSCT, hematopoietic stem cell transplantation.
Hematologic abnormalities and telomere lengths in carriers of heterozygous variants in PARN. (A-C) Pedigrees of families with segregating PARN variants. A circle represents a woman, and a square represents a man. Roman numerals indicate generations (I, II, III, IV). A slash through the circle or square indicates the person is deceased. The proband is indicated by the arrow. The variant carrier status is mut/+ for carriers and +/+ for wild type. OC indicates an individual who is an obligate carrier of the familial PARN variant. Color-shaded quarters represent different phenotypes differentiated by color: red stands for cytopenia, AA, or BMF; purple stands for ILD; blue indicates the diagnosis of a malignancy; and green marks all other TBD-associated symptoms. The exact disease/phenotype is detailed below the symbol. Pedigree numbers and the variant found in each family are all designated above the pedigree. Longitudinal hemoglobin (D) and platelet count (E) in patients pre– and post–lung transplantation. Spaghetti plots depict longitudinal blood counts from 3 PARN variant carriers (purple, red, orange) and 2 TERT variant carriers (green, blue) pre– and post–lung transplantation. The time of double-lung transplant is indicated as time point 0, and negative and positive values on the x-axis indicate the number of months before or after transplantation. (F) Peripheral blood lymphocyte telomere lengths as measured by flowFISH. Telomere length from each individual variant carrier is displayed as a red dot with his or her respective pedigree and individual ID number. Each individual’s age-adjusted telomere length is plotted compared with the testing laboratory’s population of healthy age-adjusted control subjects (in percentiles). HSCT, hematopoietic stem cell transplantation.
ILD, with a usual interstitial pneumonia pattern on imaging in all whose images were available, was diagnosed in 11 of 17 PARN P/LP variant carriers at a median age of 62.5 years (range, 49-77 years) (Table 1; supplemental Tables 4 and 5). Three PARN carriers required lung transplantation. Despite normal to high blood counts pretransplantation, patient 557 II.1 developed pancytopenia following a double-lung transplantation (Table 2; Figure 3D-E), requiring medication dose reductions, packed red blood cell transfusions, and granulocyte-colony stimulating factor. He also experienced sternal wound dehiscence, anastomosis site insufficiency, severe infections, and renal failure requiring hemodialysis. All of these complications are similar to those seen in 2 P/LP TERT carriers post–lung transplantation in this report (Table 2) and in 3 other series.27,43,44
Hematologic and other complications post-lung transplant in 3 heterozygous PARN and 2 heterozygous TERT variant carriers
| ID | Sex | Gene | Age at transplant, y | Lung transplant | Follow-up, mo | Immunosuppressants | Immunosuppressant dose reduction | CBC pretransplant | Hematologic complications | Respiratory complications | Wound healing complications | Infectious complications | CMV status D/R | CMV reactivation | Renal complications | Gastrointestinal complications | Other complications |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 557 II.1 | M | PARN | 52 | Double | 24 | Basiliximab induction, methylprednisolone, MMF, tacrolimus, leflunomide | Tacrolimus (AKI), leflunomide discontinued (pancytopenia) | WBC 9.5 × 103/μL; ANC 6.93 × 103/μL; Hb 17.7 g/dL; MCV 90.8 fL; Plts 204 × 103/μL | Hypogammaglobulinemia, anemia (∼8 g/dL, requiring RBC transfusions), thrombocytopenia (∼70-130 × 103/μL), neutropenia (∼1 × 103/μL, requiring G-CSF) | Primary graft dysfunction with lung opacity and atelectasis, bronchopleural fistula, multiple air leaks due to anastomosis insufficiency s/p multiple stents, intercostal muscle flap and pericardial wrap, chronic respiratory failure, ventilation dependency | Sternal dehiscence, anastomosis insufficiency | Sepsis (Enterococcus cloacae, Enterococcus faecalis), pulmonary infection (Staphylococcus epidermidis, rhinovirus, Candida famata, Candida guilliermondii, Candida glabrata, RSV, Pseudomonas, adenovirus, Providencia stuartii, Streptococcus agalactiae, Proteus mirabilis), gastrointestinal infection (norovirus) | D+/R– | Yes, no organ manifestation (positive BAL) | Recurrent AKI (max. creatinine 3.6 mg/dL, BUN 114 mg/dL), intermittent hemodialysis | Transaminitis, paralytic ileus | Atrial flutter, severe RV dysfunction, acute DVT, hypothyroidism, moderate encephalopathy, rectus sheath hematoma, prolonged hospitalization for 11 mo |
| 1080 III.1 | M | PARN | 53 | Double | 3 | Methylprednisolone, MMF, tacrolimus | WBC 9.4 × 103/μL; ANC 6.2 × 103/μL; Hb 13.3 g/dL; MCV 99.6 fL; Plts 262 × 103/μL | Primary graft dysfunction, left hemidiaphragm paresis | Pulmonary infection (MSSA, donor-derived) | D+/R– | Transaminitis, delayed gastric emptying | Cardiogenic shock with pulmonary edema, steroid-induced DM, actinic keratosis/seborrheic dermatitis | |||||
| 841 II.1 | M | PARN | 54 | Double | 2 | Basiliximab induction, methylprednisolone, MMF, tacrolimus | WBC 8.5 × 103/μL; ANC 5.65 × 103/μL; Hb 14.3 g/dL; MCV 87 fL; Plts 242 × 103/μL | Bilateral pleural effusion s/p pigtail insertion on left side | Wound dehiscence | D+/R– | Atrial fibrillation, acute DVT, steroid-induced DM | ||||||
| 1020 II.1 | F | TERT | 53 | Double | 39 | Basiliximab induction, methylprednisolone, MMF, tacrolimus, ATG, plasmapheresis, IVIG | MMF (neutropenia) | WBC 5.3 × 103/μL; ANC 3.02 × 103/μL; Hb 12.9 g/dL; MCV 101.6 fL; Plts 201 × 103/μL | Anemia (∼9 g/dL), thrombocytopenia (70-130 × 103/μL), neutropenia (∼1 × 103/μL, requiring G-CSF) | Right pleural effusion s/p VATS-decortication | Pulmonary infection (rhinovirus, parainfluenza), recurrent URI with Pseudomonas and coronavirus | D–/R+ | Atrial fibrillation, supraventricular tachycardia, steroid-induced DM, osteoporosis | ||||
| 669 III.1 | M | TERT | 53 | Double | 12 | Basiliximab induction, methylprednisolone, tacrolimus, azathioprine | Tacrolimus (AKI), azathioprine (neutropenia, anemia) | WBC 7.3 × 103/μL; ANC 5.54 × 103/μL; Hb 13 g/dL; MCV 101.9 fL; Plts 178 × 103/μL | Anemia (∼8 g/dL, requiring RBC transfusions), thrombocytopenia (∼100-150 × 103/μL), neutropenia (∼1.3 × 103/μL) | ARF in the setting of Nocardia sepsis and AKI | Wound dehiscence of chest wall | Pneumonia (Nocardia with possible brain involvement) | D+/R– | AKI (max. creatinine 4.5 mg/dL, BUN 94 mg/dL), intermittent hemodialysis | Transaminitis, elevated bilirubin | Atrial fibrillation, recurrent pericardial effusions, steroid-induced DM |
| ID | Sex | Gene | Age at transplant, y | Lung transplant | Follow-up, mo | Immunosuppressants | Immunosuppressant dose reduction | CBC pretransplant | Hematologic complications | Respiratory complications | Wound healing complications | Infectious complications | CMV status D/R | CMV reactivation | Renal complications | Gastrointestinal complications | Other complications |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 557 II.1 | M | PARN | 52 | Double | 24 | Basiliximab induction, methylprednisolone, MMF, tacrolimus, leflunomide | Tacrolimus (AKI), leflunomide discontinued (pancytopenia) | WBC 9.5 × 103/μL; ANC 6.93 × 103/μL; Hb 17.7 g/dL; MCV 90.8 fL; Plts 204 × 103/μL | Hypogammaglobulinemia, anemia (∼8 g/dL, requiring RBC transfusions), thrombocytopenia (∼70-130 × 103/μL), neutropenia (∼1 × 103/μL, requiring G-CSF) | Primary graft dysfunction with lung opacity and atelectasis, bronchopleural fistula, multiple air leaks due to anastomosis insufficiency s/p multiple stents, intercostal muscle flap and pericardial wrap, chronic respiratory failure, ventilation dependency | Sternal dehiscence, anastomosis insufficiency | Sepsis (Enterococcus cloacae, Enterococcus faecalis), pulmonary infection (Staphylococcus epidermidis, rhinovirus, Candida famata, Candida guilliermondii, Candida glabrata, RSV, Pseudomonas, adenovirus, Providencia stuartii, Streptococcus agalactiae, Proteus mirabilis), gastrointestinal infection (norovirus) | D+/R– | Yes, no organ manifestation (positive BAL) | Recurrent AKI (max. creatinine 3.6 mg/dL, BUN 114 mg/dL), intermittent hemodialysis | Transaminitis, paralytic ileus | Atrial flutter, severe RV dysfunction, acute DVT, hypothyroidism, moderate encephalopathy, rectus sheath hematoma, prolonged hospitalization for 11 mo |
| 1080 III.1 | M | PARN | 53 | Double | 3 | Methylprednisolone, MMF, tacrolimus | WBC 9.4 × 103/μL; ANC 6.2 × 103/μL; Hb 13.3 g/dL; MCV 99.6 fL; Plts 262 × 103/μL | Primary graft dysfunction, left hemidiaphragm paresis | Pulmonary infection (MSSA, donor-derived) | D+/R– | Transaminitis, delayed gastric emptying | Cardiogenic shock with pulmonary edema, steroid-induced DM, actinic keratosis/seborrheic dermatitis | |||||
| 841 II.1 | M | PARN | 54 | Double | 2 | Basiliximab induction, methylprednisolone, MMF, tacrolimus | WBC 8.5 × 103/μL; ANC 5.65 × 103/μL; Hb 14.3 g/dL; MCV 87 fL; Plts 242 × 103/μL | Bilateral pleural effusion s/p pigtail insertion on left side | Wound dehiscence | D+/R– | Atrial fibrillation, acute DVT, steroid-induced DM | ||||||
| 1020 II.1 | F | TERT | 53 | Double | 39 | Basiliximab induction, methylprednisolone, MMF, tacrolimus, ATG, plasmapheresis, IVIG | MMF (neutropenia) | WBC 5.3 × 103/μL; ANC 3.02 × 103/μL; Hb 12.9 g/dL; MCV 101.6 fL; Plts 201 × 103/μL | Anemia (∼9 g/dL), thrombocytopenia (70-130 × 103/μL), neutropenia (∼1 × 103/μL, requiring G-CSF) | Right pleural effusion s/p VATS-decortication | Pulmonary infection (rhinovirus, parainfluenza), recurrent URI with Pseudomonas and coronavirus | D–/R+ | Atrial fibrillation, supraventricular tachycardia, steroid-induced DM, osteoporosis | ||||
| 669 III.1 | M | TERT | 53 | Double | 12 | Basiliximab induction, methylprednisolone, tacrolimus, azathioprine | Tacrolimus (AKI), azathioprine (neutropenia, anemia) | WBC 7.3 × 103/μL; ANC 5.54 × 103/μL; Hb 13 g/dL; MCV 101.9 fL; Plts 178 × 103/μL | Anemia (∼8 g/dL, requiring RBC transfusions), thrombocytopenia (∼100-150 × 103/μL), neutropenia (∼1.3 × 103/μL) | ARF in the setting of Nocardia sepsis and AKI | Wound dehiscence of chest wall | Pneumonia (Nocardia with possible brain involvement) | D+/R– | AKI (max. creatinine 4.5 mg/dL, BUN 94 mg/dL), intermittent hemodialysis | Transaminitis, elevated bilirubin | Atrial fibrillation, recurrent pericardial effusions, steroid-induced DM |
ANC, absolute neutrophil count; AKI, acute kidney failure; ARF, acute respiratory failure; ATG, antithymocyte globulin; BAL, bronchoalveolar lavage; BUN, blood urea nitrogen; CBC, complete blood cell count; CMV, cytomegalovirus, D, donor; DM, diabetes mellitus; DVT, deep vein thrombosis; F, female; G-CSF, granulocyte-colony stimulating factor; Hb, hemoglobin; IVIG, intravenous immunoglobulin; M, male; max., maximum; MCV, mean corpuscular volume; MMF, mycophenolate mofetil; MSSA, methicillin-sensitive Staphylococcus aureus; Plts, platelets; R, recipient; RBC, red blood cell; RSV, respiratory syncytial virus; RV, right ventricular; s/p, status post; URI, upper respiratory tract infection; VATS, video-assisted thoracoscopic surgery; WBC, white blood cell count.
Family 832 presented with ILD affecting 13 members in 2 generations without any hematologic manifestations. Of the 25 family members screened, we identified a heterozygous deletion encompassing exons 1-21 of PARN (Figure 2A-C) in 9 members. All 8 family members with confirmed ILD carried the familial variant or were obligate carriers. Using a high-density microarray and quantitative real-time PCR, the deletion was found to encompass additional genes with the upstream breakpoint located between exon 4 of PLAG2G10 and exon 13 of NOMO1. This deletion is larger than 3 other intragenic multi-exon PARN deletions noted in the literature, which were all associated with serious neuropsychiatric manifestations.45 In addition, 1 healthy individual carrying a heterozygous whole-gene PARN deletion has been reported.46 Notably, none of the carriers in family 832 or 1210 had neuropsychiatric disorders. From families 832 and 1080, 13 (76%) of 17 P/LP PARN variant carriers aged >50 years have developed ILD.
Telomere length testing in P/LP TBD variant carriers
We performed peripheral blood lymphocyte telomere length measurements by flowFISH in 29 of 42 confirmed P/LP variant carriers (Table 1; supplemental Figure 5). Three of the 29 had active hematologic malignancies, and 1 (1020 II.1) was post–bilateral lung transplant at the time of measurement. The remaining carriers had normal blood counts or mild cytopenias or macrocytosis. Thirteen (45%) carriers had lymphocyte telomere lengths below the first percentile, whereas 7 (24%) and 9 (31%) carriers had telomere lengths at the first percentile to below the 10th percentile and 10th percentile or above compared with age-matched control subjects, respectively. One TERC carrier (875 III.1 who had MDS at the time of measurement) and 8 PARN variant carriers accounted for those with telomere lengths at the 10th percentile or above. Among the 12 PARN P/LP variant carriers, 2 (17%) had lymphocyte telomere lengths below the first percentile, 2 (17%) were between the first and 10th percentile, and 8 (67%) were at the 10th percentile or above (P < .01 compared with the distribution in all other P/LP variant carriers) (Figure 3F).
Usual and atypical phenotypic findings in TBD P/LP variant carriers
Twelve of the 42 P/LP variant carriers exhibited one or more features known to be associated with TBD, including reticular skin hypopigmentation, early graying of hair, liver fibrosis, skin squamous cell carcinoma, or osteopenia/osteoporosis (Table 1; supplemental Table 4). Atypical presentations included the Shwachman Diamond syndrome–like presentation of proband 775 III.1 who carried a deletion of the entire TERC gene (supplemental Figure 1). He initially presented with isolated neutropenia at 14 years of age accompanied by minor bacterial skin infections. At age 20, an evaluation revealed macrocytosis, a hypocellular bone marrow without karyotypic abnormalities, and symptoms consistent with pancreatic insufficiency, including steatorrhea, weight loss, and fat-soluble vitamin deficiency that improved with pancreatic enzyme replacement. Results of a sweat chloride test were negative. Patient 218 II.1, who carries a heterozygous LP TERC variant in the context of a strong family history of AA and ILD, had an atypical AA course. She received immunosuppressive treatment with cyclosporine and antithymocyte globulin for her AA before a TBD was diagnosed. Unexpectedly, she went into a durable complete remission for ∼15 years before relapsing. Neither a second course of cyclosporine and antithymocyte globulin nor treatment with eltrombopag was effective at relapse. Danazol led to a partial response, with a decrease in transfusion dependency. Two other patients, 875 III.1 and 684.II.1, both of whom also carry LP TERC variants, developed moderate pancytopenia that remained stable for 13 and 22+ years without treatment, suggesting that even moderate BMF may have prolonged stability in some TBD cases. The former patient eventually progressed to MDS at age 53 years, emphasizing the need for markers of progression and long-term outcome data of transplant observation in these types of cases to best guide clinical management.
Finally, inflammatory and/or autoimmune-disease related ILD phenotypes were noted in several families (Figure 4A-C). These included family 865, with all 3 family members with ILD having a chronic hypersensitivity pneumonitis pattern. Nondiagnostic autoimmune features such as positive antinuclear antibodies were noted in 4 patients with ILD, Raynaud’s phenomenon, and psoriasis were each noted in one family (Table 1; supplemental Table 4). Review of lung histology revealed diffuse lymphoplasmacytic infiltrates in 5 of 6 patients (Figure 4D), and results of bone marrow biopsies revealed prominent lymphoplasmacytic infiltrates and clusters of plasma cells and eosinophils, respectively, in 2 P/LP variant carriers (Figure 4E; supplemental Table 5). Full autoimmune serology panels were not available for most carriers. A Venn-Euler diagram (Figure 4F) shows the phenotypic distribution and overlap of hematologic, pulmonary, and autoimmune features in our confirmed P/LP variant carriers.
![Hematologic, pulmonary, and autoimmune features overlap in genetically defined TBD families. (A-C) Pedigrees of families with ILD and autoimmune features. A circle represents a woman, and a square represents a man. Roman numerals indicate generations (I, II, III). A slash through the circle or square indicates the person is deceased. The proband is indicated by the arrow. The variant carrier status is mut/+ for carriers and +/+ for wild type. The exact disease/phenotype is detailed below the symbol. The pedigree number and the variant found in this family are designated above the pedigree. (D) Lung biopsy specimen of confirmed TBD variant carrier (669.III.1) with diffuse lymphoplasmacytic infiltrates. Depicted are fibroblastic foci with a diffuse lymphoplasmacytic infiltrate (magnification ×40, hematoxylin and eosin [H&E] staining). (E) Bone marrow biopsy specimen of confirmed TBD variant carrier (669.III.1) with diffuse lymphoid aggregates. This core biopsy sample shows clusters of lymphocytic aggregates (magnification ×10, H&E staining). (F) Venn-Euler diagram of the overlap of hematologic, pulmonary, and autoimmune phenotypes observed in confirmed variant carriers. The hematologic phenotype was divided into 5 different categories: MDS/AML (dark brown), macrocytosis/macrocytic anemia (red), neutropenia (teal), thrombocytopenia (purple), and AA (peach); the pulmonary phenotype observed consisted of ILD (blue). The autoimmune overlap with ILD subtypes is highlighted in green. The number within the circles represents the number of patients with this disease/combination of phenotypes.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/19/10.1182_bloodadvances.2020001721/2/m_advancesadv2020001721f4.png?Expires=1769792960&Signature=WPyeaw4URYIFHF-JwXO3D6hMTKV5cYQcgQsZrG98frf6LUKpkzglNXUX4QY4bkdRYCDAyNiu7OxzeJzvEXhyPO13DYYn2SaElr97jHTYG0nJw38wt0lAXYmR80~Gu7gV2I-f65q34bUcghY6D5mh46s2aVywsLxFweh9rKLq2QC3tD2MbG5kiy9pgLqoOf-70I2ls50-P8bwCiQg~GaGYZLaEKFxFX4CB2KZW4~KXv2J3dCGf7EUDVWKTIK2DmnkY~SjMi8OGiKASgq0qKfPZXO5B9poniSfmnp7A5sTjMiN9uzbetEPUgnf6xiuBNcr1i1j~DPFt20b9Ps0EqFzdA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Hematologic, pulmonary, and autoimmune features overlap in genetically defined TBD families. (A-C) Pedigrees of families with ILD and autoimmune features. A circle represents a woman, and a square represents a man. Roman numerals indicate generations (I, II, III). A slash through the circle or square indicates the person is deceased. The proband is indicated by the arrow. The variant carrier status is mut/+ for carriers and +/+ for wild type. The exact disease/phenotype is detailed below the symbol. The pedigree number and the variant found in this family are designated above the pedigree. (D) Lung biopsy specimen of confirmed TBD variant carrier (669.III.1) with diffuse lymphoplasmacytic infiltrates. Depicted are fibroblastic foci with a diffuse lymphoplasmacytic infiltrate (magnification ×40, hematoxylin and eosin [H&E] staining). (E) Bone marrow biopsy specimen of confirmed TBD variant carrier (669.III.1) with diffuse lymphoid aggregates. This core biopsy sample shows clusters of lymphocytic aggregates (magnification ×10, H&E staining). (F) Venn-Euler diagram of the overlap of hematologic, pulmonary, and autoimmune phenotypes observed in confirmed variant carriers. The hematologic phenotype was divided into 5 different categories: MDS/AML (dark brown), macrocytosis/macrocytic anemia (red), neutropenia (teal), thrombocytopenia (purple), and AA (peach); the pulmonary phenotype observed consisted of ILD (blue). The autoimmune overlap with ILD subtypes is highlighted in green. The number within the circles represents the number of patients with this disease/combination of phenotypes.
Hematologic, pulmonary, and autoimmune features overlap in genetically defined TBD families. (A-C) Pedigrees of families with ILD and autoimmune features. A circle represents a woman, and a square represents a man. Roman numerals indicate generations (I, II, III). A slash through the circle or square indicates the person is deceased. The proband is indicated by the arrow. The variant carrier status is mut/+ for carriers and +/+ for wild type. The exact disease/phenotype is detailed below the symbol. The pedigree number and the variant found in this family are designated above the pedigree. (D) Lung biopsy specimen of confirmed TBD variant carrier (669.III.1) with diffuse lymphoplasmacytic infiltrates. Depicted are fibroblastic foci with a diffuse lymphoplasmacytic infiltrate (magnification ×40, hematoxylin and eosin [H&E] staining). (E) Bone marrow biopsy specimen of confirmed TBD variant carrier (669.III.1) with diffuse lymphoid aggregates. This core biopsy sample shows clusters of lymphocytic aggregates (magnification ×10, H&E staining). (F) Venn-Euler diagram of the overlap of hematologic, pulmonary, and autoimmune phenotypes observed in confirmed variant carriers. The hematologic phenotype was divided into 5 different categories: MDS/AML (dark brown), macrocytosis/macrocytic anemia (red), neutropenia (teal), thrombocytopenia (purple), and AA (peach); the pulmonary phenotype observed consisted of ILD (blue). The autoimmune overlap with ILD subtypes is highlighted in green. The number within the circles represents the number of patients with this disease/combination of phenotypes.
Discussion
Using next-generation sequencing assays, we found inherited P/LP variants in TBD genes in 16 (10%) of 153 probands at risk for a TBD due to hematologic and/or pulmonary personal and/or family history presentations. Overall, 19% of the causative variants were large deletions, highlighting the need for testing strategies that also incorporate copy number variant detection. Among the 139 with familial hematologic disorders, prevalence of TBD was 41% in those with hematologic manifestations and at least 1 case of ILD vs 3% in those with hematologic-only presentations; this suggests that ILD history is a high-yield screening question in adults with cytopenias and hematologic malignancies to help select patients for TBD genetic testing. Lymphocyte telomere lengths were at the 10th percentile or greater in 9 (31%) of 29 P/LP variant carriers, primarily accounted for by PARN carriers, among whom 67% had telomere lengths in this range. Only 24% of heterozygous PARN P/LP variant carriers had blood count abnormalities, but significant bone marrow hypocellularity observed pre–lung transplantation and severe cytopenias post–lung transplantation in 2 carriers suggest the possibility of subclinical bone marrow abnormalities, similar to findings observed for TERT, TERC, and RTEL1 P/LP variant carriers.26,27,47
Our series confirms and extends the finding that families in which hematologic disorders overlap with ILD are highly likely to have a TBD. Parry et al29 found TERT or TERC P/LP variants in 10 of 10 families with AA or hypoplastic bone marrows and ILD. Here we found that 41% of families with even mild hematologic manifestations, such as mild cytopenias or macrocytosis alone, or AA and ILD have a P/LP variant distributed among TERT, TERC, and PARN. These data suggest that hematologists should screen for a personal/family history of ILD and pulmonologists for cytopenias, macrocytosis, AA, or hematologic malignancies. All of those who screen positive for this combination warrant a multi-gene testing approach able to detect copy number variants and PARN, a gene not yet commonly included on hereditary hematologic syndrome gene panels.
In contrast, our overall yield of TBD genetic testing in families clustering hematologic disorders in our series of 3% (4 of 117) is significantly lower than the 19% (5 of 27; P = .01) observed by Holme et al24 and is more congruent with the 1% to 5% estimates in sporadic AA/MDS/AML.18,19,23 This discrepancy is likely due to differing definitions of familial hematologic disorders, with their series enriched for AA cases vs ours with familial MDS/acute leukemia or chronic cytopenia cases without AA. Thus, AA will enrich for TBD among familial hematologic disorder cases. Using our case definition, TBD genes tie RUNX1 (n = 4 [3%]) and fall behind only DDX41 (n = 7 [6%]) in accounting for the most familial hematologic disorder diagnoses. Thus, TBD genes, especially TERT, TERC, RTEL1, and PARN, are important to include on the differential of hereditary hematologic disorder evaluations.
Fifty to seventy percent of patients with familial pulmonary fibrosis and 30% to 40% of patients with dyskeratosis congenita do not have an identifiable P/LP variant in any known TBD gene.8,11,14,48 Similarly, in our series, many probands with a personal/family history of ILD, MDS/acute leukemia, and/or chronic BMF/AA did not have an identifiable P/LP TBD gene variant. For these patients, including the 9 we identified with a TBD gene variant of uncertain significance awaiting additional functional testing and segregation analyses to allow updated classification, a clinical TBD diagnosis is still possible if critically short lymphocyte telomeres and/or a classical dyskeratosis congenita phenotype are present. However, in the absence of a diagnostic phenotype, if lymphocyte telomere lengths fall within the normal range, a definitive conclusion can be difficult. These patients should be encouraged to participate in research and be followed up longitudinally to monitor for additional phenotypic features and to allow updated genetic testing as new genes are discovered. Management must be individualized in this situation, deciding whether enough evidence is present to warrant TBD-specific management of any organ pathologies that arise.49
Clinical diagnosis can also be hampered when plausible alternative etiologies are found for possible TBD-related phenotypic features. For example, autoimmune causes of combined cytopenia(s) and ILD, such as rheumatoid arthritis, are known. Recent reports have identified P/LP variants in TBD genes in populations with clinically diagnosed autoimmune disorders or, conversely, have diagnosed autoimmune diseases in patients with known TBD P/LP variants. Juge et al50 found germline P/LP RTEL1, PARN, or TERT variants in 11% of patients with rheumatoid arthritis–associated ILD. Conversely, ILD subtypes described in patients with P/LP variants in RTEL1, TERT, and TERC included autoimmune or inflammatory disease–associated subtypes as we saw in our cases.10,51,52 Although not diagnostic of overt autoimmune diseases, our findings of elevated antinuclear antibodies, Raynaud’s syndrome, and lymphoplasmacytic infiltrates in lung and bone marrow biopsy specimens similar to those found in intestinal biopsy specimens in TBD cases by others53 in confirmed TBD carriers fit well with these reports. How autoimmunity, lymphocyte telomere length, and TBD are or are not intertwined clinically or mechanistically remain to be determined.54-56 In either case, given data that immunosuppressive agents may be ineffective or even harmful in the treatment of TBD-related BMF or ILD,57 maintaining a high index of suspicion for TBD despite autoimmune features is critical.
Whereas biallelic disruption of PARN can lead to classical dyskeratosis congenita with BMF, the hematopoietic phenotype of heterozygous P/LP PARN variant carriers is less characterized.8,12 Stuart et al12 found asymptomatic blood count abnormalities, including macrocytosis (n = 3), anemia (n = 3), and thrombocytopenia (n = 1), in 6 (55%) of 11 carriers with ILD from 6 families with familial pulmonary fibrosis. Here we found macrocytosis, thrombocytopenia, or polycythemia in 4 (24%) of 17 P/LP PARN variant carriers from 5 families. One carrier had a hypocellular bone marrow with dyserythropoiesis and another had severe pancytopenia requiring transfusions, granulocyte-colony stimulating factor, and immunosuppressant dose reductions after a double lung transplant, suggesting underlying bone marrow deficits. This is similar to findings in patients with P/LP TERT, TERC, and RTEL1 variants who underwent lung transplant.26,27,47 To date, hematologic malignancies have not been reported in a heterozygous PARN P/LP variant carrier in our series or elsewhere. More data are needed to determine the full range of hematologic phenotypes and optimal peri-transplantation management.
Our study has limitations. First, the small numbers of familial pulmonary fibrosis cases and the fact that all of these were referred for a hereditary hematologic disorders evaluation may bias our estimates of the overall burden of TBD to this phenotype. However, our 21% estimate falls close to the 26% found via sequencing of TERT, TERC, PARN, and RTEL1 in 185 familial pulmonary fibrosis kindreds by Stuart et al.12 Second, our estimates of lymphocyte telomere lengths by flowFISH in P/LP variant carriers could be biased by the overall small numbers and inclusion of results from related individuals. In our series, 55% of patients with a genetically diagnosed TBD had lymphocyte telomere lengths above the first percentile. Similarly, Alder et al22 found in a larger hospital-based series of patients genetically diagnosed with TBD that 84% (49 of 58) identified at age ≥40 years and 100% (17 of 17) aged ≥61 years had lymphocyte telomere lengths above the first percentile. Telomere lengths in PARN variant carriers often fall at the 10th percentile or higher: 67% in our series, 44% (4 of 9) of all unrelated probands from our study, plus those in the published literature measured by flowFISH,12,46,58 and 43% (3 of 7) probands measured by quantitative PCR fall in this range.12 Thus, lymphocyte telomere length as the sole diagnostic test for TBD in adults, especially older adults and those carrying a heterozygous PARN variant, is challenging.
In conclusion, we found P/LP variants in TBD genes in 10% of families screened based on a personal/family history with hematologic and/or pulmonary manifestations for whom a TBD was on the differential diagnosis. Among those with hematologic manifestations, the combination of both hematologic abnormalities and ILD is the highest yield genetic testing scenario. Screening for ILD in hematology patients and hematologic abnormalities in ILD patients can target all with combined features for multi-gene testing with expected high yields. Yields for those with familial hematologic disorders without ILD are still sufficient to warrant genetic testing including TBD genes, especially TERT, TERC, PARN, and RTEL1 on panels frequently used for inherited BMF, familial MDS/acute leukemia, and familial cytopenia/macrocytosis evaluations.
All pathogenic/likely pathogenic variants and variants of uncertain significance are detailed in supplemental Methods and supplemental Tables 2 and 3 and have been deposited into ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).
Acknowledgments
The authors gratefully acknowledge all of the individuals and families who participated in this study. They are grateful to Mary Armanios from Johns Hopkins University School of Medicine for helpful discussions and for assistance with telomere length measurement data and variant interpretation.
The National Institutes of Health supported this work through the National Cancer Institute (R35CA197458) (M.-C.K., T.W., and S. Gulsuner), the National Institute of Mental Health (R01MH083989) (M.-C.K., T.W., and S. Gulsuner), and the National Heart, Lung, and Blood Institute (R03HL145253-01) (J.E.C.).
Authorship
Contribution: S.F., A.A., M.E.S., and J.E.C. designed and coordinated the study, and collected, analyzed, and interpreted the data; D.M. contributed to data collection and analysis; D.M., R.V., A.H.W.D., P.S.R., A.O., R.H.C., R.H.K., S.D.G., P.G., and L.A.G. contributed to patient recruitment; Z.L., D.d.G., H.P.S., S.D., T.W., S. Gulsuner, J.P.S., and M.-C.K. contributed to genomic screening, analysis, and interpretation of sequencing and bioinformatics data; A.N.H. and S. Gurbuxani reviewed and interpreted pathology specimens; S.F. and J.E.C. wrote the manuscript; and all authors approved the final version.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for J.E.C. is Division of Hematology, Medical Oncology, and Palliative Care, Department of Medicine, The University of Wisconsin School of Medicine and Public Health and Carbone Cancer Center, Madison, WI.
Correspondence: Jane E. Churpek, University of Wisconsin–Madison, 1111 Highland Ave, Room 4057, Madison, WI 53705; e-mail: jchurpek@wisc.edu.

