

Key Points
A patient with myelodysplastic syndrome was transplanted twice and developed clonally unrelated relapse each time in donor-derived cells.
This case supports the concept that a leukemogenic marrow environment may predispose the transplant recipient to malignant transformation.

Introduction
Donor-derived leukemia (DDL) is a rare but serious event in recipients of allogeneic hematopoietic cell transplantation (allo-HCT) performed to treat various malignancies.1-6 The incidence of DDL is estimated to be ∼0.1% among allo-HCT recipients.2,6 The pathogenesis of DDL remains unclear but may involve the interplay of factors intrinsic and extrinsic to the recipient and donor, as well as toxic effects of conditioning therapy. Different etiologies have been proposed for donor cell leukemogenesis in the recipient including transferal of undiagnosed rare malignant clones from the donor, preexisting mutations of donor progenitor cells, hostility of the recipient microenvironment, stress induced by the peritransplant treatment course because of the conditioning regimen, and reduced immune surveillance because of the posttransplant immunocompromised state. To date, several reports have documented the detection of relevant mutations in archived donor samples and others have reported diagnosis of leukemia in the donor after donation of stem cells to a recipient, supporting the transfer of a malignant clone from donor to recipient or a preexisting mutation in the donor cells leading to development of DDL in susceptible recipients, respectively.1,3 Most DDL cases report no emergence of leukemia in the donor after transplantation, and in a subset of these cases, examination of donor bone marrow (BM) demonstrated no evidence of occult malignant clones.2,4 However, follow-ups are relatively short at the time of publication, and future leukemia arising in the donor cannot be eliminated.
Although there have been numerous reports of DDL, to our knowledge, there has never been a report of multiple DDLs in an individual patient. Here, we report a novel case of a patient who developed 2 distinct DDLs after allo-HCT using 2 different donors for treatment of a myeloid malignancy (Figure 1).
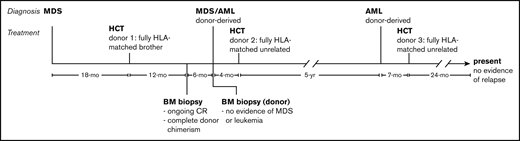
Timeline of diagnosis and treatment. The patient was initially diagnosed with MDS and subsequently donor-derived MDS/AML and donor-derived AML after allo-HCT from 2 different donors. She is currently 24 months after allo-HCT from a third donor and shows no evidence of disease. CR, complete response.
Timeline of diagnosis and treatment. The patient was initially diagnosed with MDS and subsequently donor-derived MDS/AML and donor-derived AML after allo-HCT from 2 different donors. She is currently 24 months after allo-HCT from a third donor and shows no evidence of disease. CR, complete response.
Case description
A previously healthy 60-year-old woman was diagnosed with myelodysplastic syndrome (MDS) after presenting with pancytopenia (initial white blood cell count: 2 × 103/μL; hemoglobin: 9 g/dL; platelet count: 81 × 103/μL). BM biopsy showed 3% blasts and multilineage dysplastic changes. Conventional cytogenetics showed 46,XX,del(7)(q11.2q32),del(20)(q11.2q13.1)[20] (Figure 2A). Her exposure history was unremarkable because she worked in a law firm, never smoked, and only consumed alcohol socially. She had no family history of malignancies besides a grandfather who was a smoker and died of lung cancer. Her cell counts improved after treatment with 3 cycles of azacitidine. Eighteen months later, she underwent allo-HCT from her fully HLA-matched 54-year-old brother after fludarabine and melphalan conditioning. She engrafted, achieving complete donor chimerism by day 30 after transplant. She developed limited skin graft-versus-host disease (GVHD) that responded to low-dose steroids, and immunosuppressive medications were discontinued 10 months after transplant. Her BM biopsy at 1 year after transplant was consistent with ongoing remission and complete donor chimerism. Cytogenetics showed normal male karyotype in all metaphases, and fluorescence in situ hybridization studies were negative for del(7q) or del(20q)/monosomy 20.
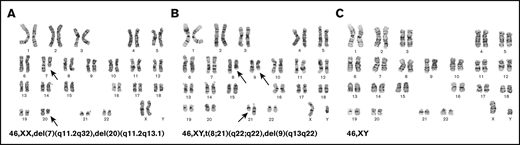
Cytogenetic abnormalities identified during the course and treatment of the disease. (A) At diagnosis, the female patient was identified to have karyotypic abnormalities consistent with MDS with deletion 7q and del 20q. (B) After allogeneic stem cell transplantation, she was found to have a donor-derived AML with t(8;21). (C) After her second allogeneic stem cell transplantation, she was found to have another AML with normal karyotype.
Cytogenetic abnormalities identified during the course and treatment of the disease. (A) At diagnosis, the female patient was identified to have karyotypic abnormalities consistent with MDS with deletion 7q and del 20q. (B) After allogeneic stem cell transplantation, she was found to have a donor-derived AML with t(8;21). (C) After her second allogeneic stem cell transplantation, she was found to have another AML with normal karyotype.
Eighteen months after transplant, she presented with pancytopenia. BM biopsy showed high-grade MDS with 12% blasts and dysplastic changes. Cytogenetics showed 46,XY,t(8;21)(q22;q22)[2], 45,sl,-Y[7], 46,sl,del(9)(q13q22)[2], 46,XY[10] (Figure 2B).
Because the core-binding factor translocation was present, she was treated using acute myeloid leukemia (AML) therapies. Analysis of marrow engraftment by quantitative polymerase chain reaction showed 99.91% donor DNA. The donor was re-evaluated, and his blood counts were normal. He underwent BM biopsy, which showed normocellular marrow without evidence of leukemia or MDS. Cytogenetics showed normal male karyotype in all metaphases.
The patient was induced using the 7+3 regimen and achieved complete remission, after which she received a cycle of high-dose cytarabine consolidation. She underwent a second allo-HCT from a fully HLA-matched, 32-year-old unrelated male donor after clofarabine and melphalan conditioning and developed mild chronic GVHD involving the mouth and eyes.
Five years after the second transplant, she developed thrombocytopenia and circulating blasts. BM biopsy showed AML with 50% blasts. Cytogenetics showed normal male karyotype in all metaphases (Figure 2C), and fluorescence in situ hybridization studies including core-binding factor were negative. Her marrow cells were predominantly (99%) derived from the unrelated donor as confirmed by quantitative polymerase chain reaction. She was reinduced with 7+3, achieved complete remission, and received decitabine consolidation. She underwent a third allo-HCT from a different fully HLA-matched unrelated donor after fludarabine and melphalan conditioning 7 months after her AML diagnosis. She developed skin GVHD after transplant that eventually resolved. She is currently 24 months after transplant and shows no evidence of relapse.
Discussion
DDL is a rare complication of allo-HCT.2 Previous reports have described a single DDL in an individual recipient, whereas we describe an individual who developed 2 distinct DDLs from 2 donors. Each instance of DDL raises the question of etiology: was DDL triggered by pre-existing factors in the donor product or by a leukemogenic microenvironment in the recipient? If the donor subsequently develops a clonally related leukemia, then that would support a preexisting tendency of the donated cells to transform. If the donor does not develop leukemia, possible alternatives include (1) DDL was caused by a preexisting mutation in the donor cells, but not enough time has elapsed for the donor to acquire a second leukemogenic mutation, or (2) DDL was triggered by a hostile leukemogenic microenvironment in the recipient that also caused the original leukemia, peritransplant factors (ie, conditioning regimens), or the posttransplant immunosuppressive state. The observation of DDL in recipients of cord blood further suggests that insults to BM stroma either from previous chemotherapy or peritransplant factors can contribute to leukemia development.7-10 Nonetheless, DDL may result from a combination of donor- and recipient-related factors.
The BM microenvironment likely plays a key role in maintaining homeostasis of hematopoietic stem cells.11 Preclinical studies have shown associations between disruption and alteration of cell populations in the marrow microenvironment (mesenchymal stem cells, stromal cells, endothelial cells, adipocytes, osteogenic cells, osteoblasts, etc), with the process of leukemia initiation, progression, or development of chemotherapy resistance.12-15 Therefore, if a patient has defective cells in the BM microenvironment that are predisposed to induce or allow leukemic transformation, replacing the hematopoietic stem cells through allo-HCT may not be sufficient to prevent recurrent DDL.
Peritransplant factors including treatment with alkylator- or radiation-based conditioning, in vivo T-cell depletion, or development of significant GVHD may result in impaired immune surveillance and increased risk of DDL.6,8 The disappearance of donor-derived abnormal clones after transplant on reconstitution of the recipient immune system has been reported and supports a role for immune surveillance.16,17 Although our patient had an alkylator (melphalan) as part of all 3 conditioning regimens, she had no radiation or T-cell depletion, and no significant GVHD occurred.
Here we present the first reported instance of a patient who developed 2 clonally distinct myeloid DDLs after 2 allo-HCTs from a related and an unrelated donor. To our knowledge, multiple DDLs in a single patient have not been reported before in the literature. This case strongly supports the notion that a leukemogenic microenvironment in the alloHCT recipient induces DDL. This is the most likely scenario here because the recipient developed 2 different leukemias from 2 different donors; the original donor was examined and did not have any detectable abnormalities and the second transplant was performed using cells from a young (32-year-old) healthy donor with a low risk of clonal hematopoiesis of indeterminate potential.18 The fact that the original diagnosis for this patient was MDS supports our theory that the leukemogenic milieu engendered recurrent DDL.
For data, please contact the corresponding author at sforman@coh.org.
Acknowledgments
The authors thank Mary Clark for editing and advice on presentation of this manuscript and Joyce Murata-Collins for providing the cytogenetic images.
Authorship
Contribution: S.J.F. and P.T.C. were the treating physicians for this case presentation; and I.A., P.T.C., J.Y.S., and S.J.F. wrote the manuscript and reviewed and approved the final version before submission.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Stephen J. Forman, Department of Hematology/HCT, City of Hope Medical Center, 1500 E. Duarte Rd, Duarte, CA 91010; e-mail: sforman@coh.org.

