

Key Points
Platelet CD36 signaling promotes hydrogen peroxide–mediated oxidative cysteine modification on Src family kinases.
Cysteine sulfenylation is important for proaggregatory and procoagulant platelet functions by oxLDL/CD36.

Abstract
Arterial thrombosis in the setting of dyslipidemia promotes clinically significant events, including myocardial infarction and stroke. Oxidized lipids in low-density lipoproteins (oxLDL) are a risk factor for athero-thrombosis and are recognized by platelet scavenger receptor CD36. oxLDL binding to CD36 promotes platelet activation and thrombosis by promoting generation of reactive oxygen species. The downstream signaling events initiated by reactive oxygen species in this setting are poorly understood. In this study, we report that CD36 signaling promotes hydrogen peroxide flux in platelets. Using carbon nucleophiles that selectively and covalently modify cysteine sulfenic acids, we found that hydrogen peroxide generated through CD36 signaling promotes cysteine sulfenylation of platelet proteins. Specifically, cysteines were sulfenylated on Src family kinases, which are signaling transducers that are recruited to CD36 upon recognition of its ligands. Cysteine sulfenylation promoted activation of Src family kinases and was prevented by using a blocking antibody to CD36 or by enzymatic degradation of hydrogen peroxide. CD36-mediated platelet aggregation and procoagulant phosphatidylserine externalization were inhibited in a concentration-dependent manner by a panel of sulfenic acid–selective carbon nucleophiles. At the same concentrations, these probes did not inhibit platelet aggregation induced by the purinergic receptor agonist adenosine diphosphate or the collagen receptor glycoprotein VI agonist collagen-related peptide. Selective modification of cysteine sulfenylation in vivo with a benzothiazine-based nucleophile rescued the enhanced arterial thrombosis seen in dyslipidemic mice back to control levels. These findings suggest that CD36 signaling generates hydrogen peroxide to oxidize cysteines within platelet proteins, including Src family kinases, and lowers the threshold for platelet activation in dyslipidemia.
Introduction
Dyslipidemia is a risk factor for clinically significant thrombotic events that are the leading causes of death by cardiovascular disease.1 Thrombosis in this context is mediated in part by heightened platelet reactivity facilitated by circulating oxidized lipids present in oxidized low-density lipoprotein particles (oxLDL).2 Current antiplatelet agents are effective at preventing platelet activation; however, these agents have limited utility due to risk of bleeding complications and incomplete efficacy in preventing recurrent thrombotic events.3
Scavenger receptor CD36 is highly expressed on the platelet surface4 and recognizes specific oxidized lipid motifs present on oxLDL particles.2 CD36 lowers the threshold for platelet activation by multiple signaling pathways5-7 through recruitment and activation of Src family kinases (SFK)5 and generation of reactive oxygen species (ROS).6,8,9 ROS generation promotes MAPK extracellular signal-regulated kinase 5 (ERK5) activation, and this signaling interacts with the collagen receptor glycoprotein VI (GPVI) pathway to amplify platelet activation and procoagulant activity.10 In addition to the direct role of CD36 in platelet activation, CD36 signaling blunts inhibitory pathways,8,9,11 thus indirectly promoting platelet activation. Furthermore, genetic studies have identified single nucleotide polymorphisms on CD36 that associate with CD36 surface expression levels12 and are linked to increased risk for coronary artery disease and myocardial infarction.13
The mechanisms through which ROS promote redox-sensitive signaling remain poorly understood.14 Although ROS modify nearly all cellular components, cysteines are particularly sensitive.15 Cysteines are oxidized by hydrogen peroxide (H2O2) to sulfenic acids, a posttranslational modification important in signaling.16 Further oxidation generates sulfinylated and sulfonylated cysteines that are believed to be irreversible17 (supplemental Figure 1). Sulfenic acids are a central hub for oxidative cysteine modifications and can be modified and detected by carbon nucleophiles.17-19 The importance of cysteine sulfenylation in thrombosis and hemostasis is largely unknown and could pinpoint redox mechanisms in conditions associated with oxidant stress.
Superoxide is generated by platelet CD36 signaling through reduced NADP (NADPH) oxidase6,9 and promotes activation of ERK5, which then induces 2 phenotypes: (1) platelet activation and aggregation for thrombosis6 ; and (2) caspase-dependent procoagulant phosphatidylserine (PSer) externalization for fibrin deposition in vivo.10 Because superoxide is a one-electron oxidant that does not directly oxidize cysteines,20 other oxidants derived from superoxide are likely the key effectors regulating CD36 signaling. We report that CD36 signaling generates H2O2, which promotes cysteine sulfenylation. In addition, SFK are sulfenylated by H2O2, which promotes activation of the kinase. Modifying sulfenylation with carbon nucleophiles prevented platelet activation, aggregation, and procoagulant PSer externalization by oxLDL/CD36 while maintaining these same processes by “classic” physiologic activators. These findings identify a targetable redox-dependent mechanism by which CD36 promotes thrombosis in dyslipidemic conditions.
Methods
H2O2 quantification
Washed human platelets (6 × 105/µL) were loaded with 10 µM coumarin boronic acid (CBA) for 15 minutes at room temperature in the dark followed by treatment with 1 µg/mL FA6 anti-CD36 or nonimmune immunoglobulin G (IgG) or 1000 U/mL denatured or functional polyethylene glycol (PEG)–catalase for 15 minutes. Before activating platelets with 50 µg/mL LDL or oxLDL for 60 minutes, 1 mM CaCl2/MgCl2 was added. Platelets were pelleted by centrifugation at 700g for 10 minutes, and pelleted samples were processed for high-performance liquid chromatography (HPLC) analysis as previously described21 (supplemental Methods).
Cysteine sulfenylation
Washed human platelets (3 × 105/µL) were loaded with 1 mM alkyne-containing benzothiazine-based probe (BTD-alkyne) for 15 minutes at room temperature. Then, 1 mM CaCl2/MgCl2 was added before activating platelets with phosphate-buffered saline (PBS), 50 µg/mL LDL or oxLDL, or treating with 0.55 mM glucose and 1 U/mL glucose oxidase up to 1 hour at room temperature. Platelets were lysed with radioimmunoprecipitation assay buffer followed by click chemistry using 200 µM biotin-PEG3-azide, 1 mM tris(2-carboxyethyl)phosphine, 0.1 mM tris([1-benzyl-1H-1,2,3-triazol-4-yl]methyl)amine, and 1 mM Cu(II)SO4 to covalently link biotin to the alkyne (supplemental Figure 1). Reactions were rocked for 15 minutes at 37°C followed by 45 minutes at room temperature. Samples were flash frozen for 10 minutes in liquid nitrogen and then dried on a SpeedVac. Samples were resuspended in PBS with 1% wt/vol sodium dodecyl sulfate (SDS) followed by Laemmli sample buffer and incubated for 10 minutes at 100°C before separation on a TGX SDS-polyacrylamide gel (Bio-Rad). Proteins were then transferred onto nitrocellulose, and biotin was detected by using Vectastain ABC with enhanced chemiluminescence reagents.
In some experiments, SFK were immunoprecipitated by using an anti-Src monoclonal antibody with Protein A/G agarose beads. Protein-antibody complexes on A/G agarose were subjected to click chemistry followed by detection of biotin using Vectastain ABC and standard enhanced chemiluminescence reagents.
Detection of oxidative cysteine modification on SFK
Washed human platelets (3 × 105/µL) were stimulated with 50 µg/mL oxLDL or 500 µM H2O2 up to 60 minutes after adding 1 mM CaCl2/MgCl2. Platelets were lysed with CelLytic M lysis buffer (Sigma-Aldrich) on ice followed by thiol alkylation with 5 µM 5-iodoacetamidofluorescein (5-IAF) for 15 minutes. Cellular debris was eliminated by centrifuging at 13 200 rpm for 10 minutes at 4°C. Supernatant was collected and precleared with washed A/G beads for 1 hour at 4°C. SFK were immunoprecipitated from precleared lysate by an anti-Src monoclonal antibody overnight. Antibody-protein complexes were washed with ice-cold lysis buffer and resuspended in Laemmli sample buffer. Samples were run on a TGX SDS-polyacrylamide gel followed by immunoblotting for fluorescein or total Src as a loading control.
Intravital microscopy of in vivo thrombosis
The ferric chloride (FeCl3)–induced carotid artery thrombosis model was performed as previously described.22-24 Ten- to 12-week-old female wild-type C57BL/6 mice on chow or 4 weeks of a Western diet (TD.88137) were injected intraperitoneally with dimethyl sulfoxide (DMSO) or BTD 3 hours before FeCl3 injury was initiated. The end points were set as: (1) blood flow ceased for >30 seconds; or (2) if blood flow cessation was not seen at 30 minutes, then 30 minutes was assigned to that mouse for statistical analysis. For the laser ablation cremaster artery thrombosis model, 8- to 12-week-old male wild-type C57BL/6 mice were treated intraperitoneally with DMSO or 25 mg/kg BTD 3 hours before laser ablation. Laser ablation was performed after intravenous injection of PBS for the control cohort. Laser ablation was then performed after 2.5 mg/kg oxLDL was injected intravenously.25,26 BTD was prepared in 20% vol/vol cremophore in PBS. Mice receiving 20% v/v cremophore in PBS containing DMSO were used as controls. Detailed information is provided in the supplemental Methods.
Platelet aggregometry, flow cytometry, and statistical analysis
Detailed information on platelet aggregometry, flow cytometry, and statistical analysis is provided in the supplemental Methods.
Results
OxLDL promotes H2O2 generation in platelets
CD36 signaling generates superoxide through NADPH oxidase.6,9 Superoxide dismutates to H2O2 spontaneously or catalytically through superoxide dismutase.20 H2O2 functionally promoted >50% of platelet aggregation by oxidized lipids6 ; however, quantitative measurements of H2O2 generation by oxLDL had not been reported. H2O2 levels in platelets were quantified by using a boronate-based probe for peroxides, CBA.27 CBA is nonfluorescent, whereas the peroxide-dependent oxidation product, 7-hydroxycoumarin (COH), exhibits 100-fold more fluorescence at the same concentration (Figure 1A). Consistent with our hypothesis, oxLDL promoted COH accumulation, whereas control LDL yielded a similar COH level as buffer treatment (Figure 1B). OxLDL treatment consistently showed a >20% increase in COH accumulation compared with LDL when platelets were stimulated with increasing oxLDL concentrations (Figure 1C). Furthermore, sensitizing platelets with oxLDL before stimulating with the GPVI agonist collagen-related peptide revealed significantly augmented COH formation relative to either collagen-related peptide or oxLDL alone (Figure 1D). This synergy between oxLDL and collagen-related peptide is consistent with previous data showing cross talk between platelet CD36 and GPVI signaling.10 Platelets stimulated with thrombin exhibited modest increased levels of COH formation (P = .06) (supplemental Figure 2A), which may be consistent with a role for thrombin in ROS generation.28,29
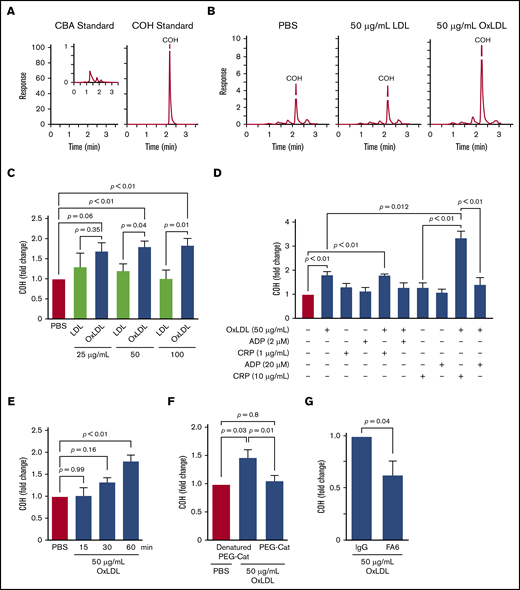
Platelet CD36 signaling generates H2O2. (A) HPLC chromatograms of 5 µM CBA or COH standards. (B) Representative HPLC chromatograms of COH accumulation by oxLDL-, LDL-, and PBS-treated platelets. (C) OxLDL in a concentration-dependent manner promotes H2O2 accumulation. Human platelets (6 × 108/mL) were preloaded with 10 µM CBA followed by stimulation with 25, 50, or 100 µg/mL lipoprotein for 60 minutes. Platelets were lysed, and COH was extracted for HPLC analyses. (D) oxLDL/CD36 signaling synergistically promotes H2O2 generation by the GPVI pathway. Human platelets (6 × 108/mL) were preloaded with 10 µM CBA followed by pretreatment with or without 50 µg/mL oxLDL for 60 minutes before further activation with 2 µM or 20 µM ADP, and/or 1 µg/mL or 10 µg/mL collagen-related peptide (CRP) for 15 minutes. Platelets were lysed, and COH was extracted for HPLC analyses. (E) oxLDL promotes H2O2 accumulation over time. Human platelets (6 × 108/mL) were preloaded with 10 µM CBA followed by stimulation with PBS or 50 µg/mL oxLDL for up to 60 minutes. Platelets were lysed, and COH was extracted for HPLC analyses. CBA oxidation by oxLDL signaling is through CD36 and H2O2. Human platelets (6 × 108/mL) were preloaded with 1000 U/mL denatured (“boiled”) PEG-catalase or functional PEG-catalase (PEG-Cat) (F) or pretreated with 1 µg/mL IgG or FA6-152 monoclonal antibody (FA6) (G) for 15 minutes, followed by 50 µg/mL oxLDL stimulation for 60 minutes. P values were determined by 1-way analysis of variance with Dunnett’s post hoc analysis (C,E,F), the paired Student t test (G), and 1-way analysis of variance with Tukey’s post hoc analysis (D). P = .66 for 25 µg/mL LDL vs PBS; P = .88 for 50 µg/mL LDL vs PBS (C); and P > .99 for 100 µg/mL LDL vs PBS. N ≥ 4 separate donors (C-D,F), and 3 separate donors (E,G). Data are expressed as mean ± SEM.
Platelet CD36 signaling generates H2O2. (A) HPLC chromatograms of 5 µM CBA or COH standards. (B) Representative HPLC chromatograms of COH accumulation by oxLDL-, LDL-, and PBS-treated platelets. (C) OxLDL in a concentration-dependent manner promotes H2O2 accumulation. Human platelets (6 × 108/mL) were preloaded with 10 µM CBA followed by stimulation with 25, 50, or 100 µg/mL lipoprotein for 60 minutes. Platelets were lysed, and COH was extracted for HPLC analyses. (D) oxLDL/CD36 signaling synergistically promotes H2O2 generation by the GPVI pathway. Human platelets (6 × 108/mL) were preloaded with 10 µM CBA followed by pretreatment with or without 50 µg/mL oxLDL for 60 minutes before further activation with 2 µM or 20 µM ADP, and/or 1 µg/mL or 10 µg/mL collagen-related peptide (CRP) for 15 minutes. Platelets were lysed, and COH was extracted for HPLC analyses. (E) oxLDL promotes H2O2 accumulation over time. Human platelets (6 × 108/mL) were preloaded with 10 µM CBA followed by stimulation with PBS or 50 µg/mL oxLDL for up to 60 minutes. Platelets were lysed, and COH was extracted for HPLC analyses. CBA oxidation by oxLDL signaling is through CD36 and H2O2. Human platelets (6 × 108/mL) were preloaded with 1000 U/mL denatured (“boiled”) PEG-catalase or functional PEG-catalase (PEG-Cat) (F) or pretreated with 1 µg/mL IgG or FA6-152 monoclonal antibody (FA6) (G) for 15 minutes, followed by 50 µg/mL oxLDL stimulation for 60 minutes. P values were determined by 1-way analysis of variance with Dunnett’s post hoc analysis (C,E,F), the paired Student t test (G), and 1-way analysis of variance with Tukey’s post hoc analysis (D). P = .66 for 25 µg/mL LDL vs PBS; P = .88 for 50 µg/mL LDL vs PBS (C); and P > .99 for 100 µg/mL LDL vs PBS. N ≥ 4 separate donors (C-D,F), and 3 separate donors (E,G). Data are expressed as mean ± SEM.
COH accumulation kinetics were then determined by stimulating platelets with oxLDL up to 60 minutes, and we found that COH accumulated in a time-dependent manner (Figure 1E) consistent with the previously described superoxide accumulation kinetics by CD36.6 Given that CBA can be oxidized by a variety of peroxides,27,30 we confirmed that H2O2 is the ROS responsible for COH formation using PEG-catalase, an enzyme that degrades H2O2 to water and dioxygen. In the presence of PEG-catalase, the COH level formed by oxLDL treatment decreased to the level observed with unstimulated buffer-treated platelets (Figure 1F), suggestive of an H2O2-dependent mechanism. We then determined CD36 dependency by measuring COH formation in the presence of the CD36-blocking monoclonal antibody FA6-152 (FA6). oxLDL-stimulated COH formation was decreased by >75% by the antibody compared with conditions with a nonimmune IgG control (Figure 1G). In addition, a specific oxidized lipid, 1-(palmitoyl)-2-(5-keto-6-octene-dioyl)phosphatidylcholine (KODia-PC), recognized by CD36 also showed an increase in COH fluorescence relative to unoxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphatidylcholine lipids (supplemental Figure 2B), consistent with KODia-PC promoting CD36-dependent ROS generation.8 The nonoxidized nonlipid CD36 ligand myeloid-related protein 14 (MRP-14) also induced an ∼30% increase in COH fluorescence within the first 5 minutes of stimulation, which was not observed in the presence of the CD36-blocking FA6 antibody (supplemental Figure 2C). These data indicate that oxLDL promote H2O2 generation in platelets through a CD36-dependent pathway.
H2O2 generation by CD36 signaling promotes cysteine sulfenylation
H2O2 promotes oxidation of redox-sensitive proteins important in modulating platelet function.14 To test the hypothesis that H2O2 oxidizes cysteines in platelets, an alkyne-containing benzothiazine-based probe (BTD-alkyne) was used to quantify cysteine sulfenylation.15 The alkyne moiety is amenable to copper-mediated azide/alkyne cycloaddition (“click chemistry”), which allows for detection of BTD-alkyne labeling by any reporter harboring an azide (supplemental Figure 1). As a positive control, platelets were treated with glucose/glucose oxidase to generate a continuous flux of H2O2. H2O2 flux increased BTD-alkyne incorporation into platelet proteins, detected by click chemistry with biotin-PEG3-azide, compared with untreated conditions (Figure 2A). Supporting our hypothesis, oxLDL promoted rapid (within 5 minutes) BTD-alkyne incorporation onto platelet proteins. Maximal BTD-alkyne incorporation occurred within 15 minutes of stimulation and decreased back to buffer-treated levels by 60 minutes. oxLDL-dependent BTD-alkyne incorporation was concentration dependent, with a 2.5-fold increase compared with control LDL seen at 50 µg/mL (Figure 2B). Importantly, BTD-alkyne omission in assays containing glucose/glucose oxidase showed no detectable biotin, indicating the signal observed required ligation of BTD-alkyne–labeled proteins to biotin-PEG3-azide. Treatment with a nonoxidized CD36 ligand, the calcium-binding S100 family member MRP-14,31 also increased sulfenylation (Figure 2D). Both oxLDL- and MRP-14–induced sulfenylation were decreased in the presence of the CD36-blocking antibody FA6-152, consistent with a CD36-dependent mechanism (Figure 2C-D). Furthermore, platelet sulfenylation by oxLDL was prevented by PEG-catalase (Figure 2E), indicating a direct role for H2O2 in mediating CD36-dependent cysteine sulfenylation.
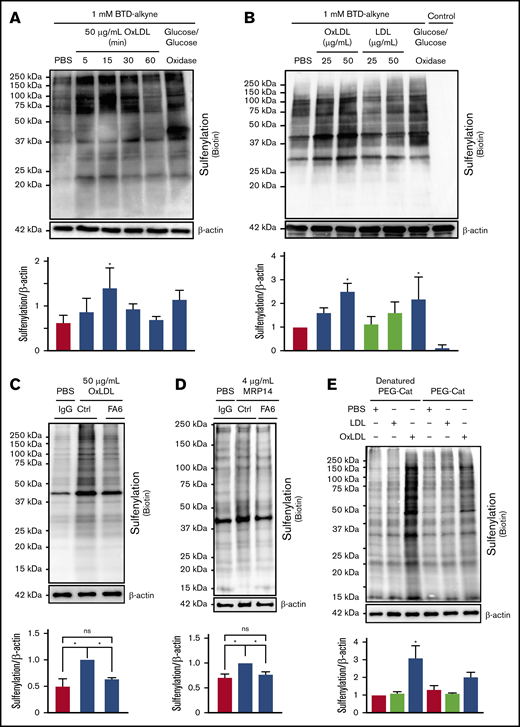
OxLDL/CD36–mediated H2O2generation promotes protein sulfenylation in platelets. OxLDL promotes protein sulfenylation over time (A) and in a concentration-dependent manner (B). (A-B) Human platelets (3 × 108/mL) were pretreated with 1 mM alkyne-containing benzothiazine-based probe (BTD-alkyne) for 15 minutes, followed by stimulation with 50 µg/mL oxLDL or treatment with 0.55 mM glucose and 1 U/mL glucose oxidase up to 60 minutes for a continuous low flux of H2O2 (A) or with increasing concentrations of LDL or oxLDL for 15 minutes (B). A control without BTD-alkyne was incorporated in panel B. Cysteine sulfenylation by oxLDL (C) and nonoxidized CD36 ligand MRP-14 (D) were decreased by the CD36-blocking antibody. Platelets were pretreated with 1 µg/mL nonimmunizing control antibody or the CD36-blocking monoclonal antibody FA6-152 (FA6) for 15 minutes in the presence of 0.1 mM BTD-alkyne followed by platelet activation with 50 µg/mL oxLDL (C) or 4 µg/mL MRP-14 (D) for 15 minutes. (E) Cysteine sulfenylation by oxLDL/CD36 signaling is through an H2O2-dependent mechanism. Platelets were pretreated with 1000 U/mL denatured PEG-catalase or functional PEG-catalase for 15 minutes in the presence of 0.1 mM BTD-alkyne followed by platelet activation with 50 µg/mL LDL or oxLDL for 15 minutes. In all panels, platelets were lysed with radioimmunoprecipitation assay buffer followed by modification of the alkyne of BTD-alkyne with biotin-PEG3-azide via azide-alkyne cycloaddition (click chemistry). P values were determined by 1-way analysis of variance with Dunnett’s post hoc analysis. *P < .05 compared with buffer treatment (A-B,E). *P < .05 with their respective comparisons (C-D). N = 5 separate donors (A), N = 4 separate donors (B), N = 4 separate donors (C-D), and N = 3 separate donors (E). Data are expressed as mean ± SEM. ns, not significant.
OxLDL/CD36–mediated H2O2generation promotes protein sulfenylation in platelets. OxLDL promotes protein sulfenylation over time (A) and in a concentration-dependent manner (B). (A-B) Human platelets (3 × 108/mL) were pretreated with 1 mM alkyne-containing benzothiazine-based probe (BTD-alkyne) for 15 minutes, followed by stimulation with 50 µg/mL oxLDL or treatment with 0.55 mM glucose and 1 U/mL glucose oxidase up to 60 minutes for a continuous low flux of H2O2 (A) or with increasing concentrations of LDL or oxLDL for 15 minutes (B). A control without BTD-alkyne was incorporated in panel B. Cysteine sulfenylation by oxLDL (C) and nonoxidized CD36 ligand MRP-14 (D) were decreased by the CD36-blocking antibody. Platelets were pretreated with 1 µg/mL nonimmunizing control antibody or the CD36-blocking monoclonal antibody FA6-152 (FA6) for 15 minutes in the presence of 0.1 mM BTD-alkyne followed by platelet activation with 50 µg/mL oxLDL (C) or 4 µg/mL MRP-14 (D) for 15 minutes. (E) Cysteine sulfenylation by oxLDL/CD36 signaling is through an H2O2-dependent mechanism. Platelets were pretreated with 1000 U/mL denatured PEG-catalase or functional PEG-catalase for 15 minutes in the presence of 0.1 mM BTD-alkyne followed by platelet activation with 50 µg/mL LDL or oxLDL for 15 minutes. In all panels, platelets were lysed with radioimmunoprecipitation assay buffer followed by modification of the alkyne of BTD-alkyne with biotin-PEG3-azide via azide-alkyne cycloaddition (click chemistry). P values were determined by 1-way analysis of variance with Dunnett’s post hoc analysis. *P < .05 compared with buffer treatment (A-B,E). *P < .05 with their respective comparisons (C-D). N = 5 separate donors (A), N = 4 separate donors (B), N = 4 separate donors (C-D), and N = 3 separate donors (E). Data are expressed as mean ± SEM. ns, not significant.
oxLDL/CD36 signaling promotes H2O2-mediated sulfenylation and activation of SFK
We next investigated the candidate proteins that are signaling effectors in the CD36 pathway. The SFK Fyn and Lyn are recruited to CD36 as a consequence of ligand binding and are signal transducers for the receptor.5,32 Of relevance, C185 and C277 in Src were oxidized in cancer cells under conditions of elevated ROS generation to maintain kinase activation.33 In platelets, SFK modification by oxidants had not been tested.
To test the hypothesis that SFK cysteines are oxidatively modified by oxLDL/CD36 signaling, we used an electrophilic (5-IAF) probe that alkylates free cysteines; cysteine oxidation results in decreased alkylation and thus decreased fluorescein signal. We previously used 5-IAF to show that CD36 promotes oxidative cysteine modification of phosphotyrosine phosphatase SHP-2 in macrophages34 and SHP-1 in microvascular endothelial cells.35 Platelets were treated with oxLDL or 500 µM H2O2 (as a positive control), and SFK were immunoprecipitated and blotted for 5-IAF. H2O2 treatment resulted in 90% decreased 5-IAF alkylation compared with control (Figure 3A). After oxLDL stimulation, a time-dependent decrease in 5-IAF alkylation was observed with a 75% decrease within 15 minutes. These data inversely correlate with the increase in COH fluorescence upon oxLDL treatment observed in Figure 1E.
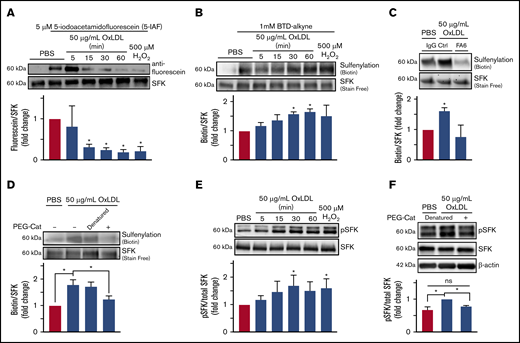
OxLDL-CD36 signaling promotes SFK cysteine sulfenylation. (A) oxLDL promotes cysteine modification of SFK in platelets. Washed human platelets (3 × 108/mL) were stimulated with buffer or 50 µg/mL oxLDL up to 60 minutes or with 500 µM H2O2 as a positive control for 15 minutes. Platelets were lysed, and unmodified cysteine thiols were alkylated with 5 µM of 5-IAF; oxidized thiols are unable to be alkylated by 5-IAF. SFK were immunoprecipitated and washed, and 5-IAF was detected by an anti-fluorescein antibody. Total SFK were detected by an anti-Src antibody. (B) oxLDL promotes SFK cysteine sulfenylation over time. (C) SFK cysteine sulfenylation by oxLDL is CD36 dependent. (D) H2O2 is the ROS downstream of CD36 that sulfenylates SFK. Washed human platelets (3 × 108/mL) were pretreated with 1 mM BTD-alkyne (B) in the presence of 1 µg/mL CD36-blocking FA6-152 (FA6) or control IgG antibody (C), or with 1000 U/mL of denatured or functional PEG-catalase (PEG-Cat) (D). The platelets were then stimulated with 50 µg/mL oxLDL up to 60 minutes (B-D), or with 500 µM H2O2 as a positive control for 15 minutes (B). Levels of SFK immunoprecipitated were assessed by UV-dependent stain-free imaging of the Bio-Rad TGX gels. (E) oxLDL/CD36 signaling promotes activation of SFK over time. (F) SFK activation by CD36 is H2O2 dependent. Washed human platelets (3 × 108/mL) were stimulated with 50 µg/mL oxLDL up to 60 minutes or with 500 µM H2O2 as a positive control (E) or with 50 µg/mL oxLDL following pretreatment with 1000 U/mL functional or denatured PEG-catalase (PEG-Cat) (F) for 15 minutes. The cells were lysed with radioimmunoprecipitation assay buffer, and phosphorylated SFK (Y416) were detected by immunoblotting. Total SFK was detected by an anti-Src antibody. P values were determined by 1-way analysis of variance with Dunnett’s posthoc analysis (A-C,E) and by 1-way analysis of variance with Tukey’s post hoc analysis (D,F). N = 3 separate donors (A-F). *P < .05 compared with buffer treatment (A-C,E). *P < .05 with their respective comparisons (D,F). Data are expressed as mean ± SEM.
OxLDL-CD36 signaling promotes SFK cysteine sulfenylation. (A) oxLDL promotes cysteine modification of SFK in platelets. Washed human platelets (3 × 108/mL) were stimulated with buffer or 50 µg/mL oxLDL up to 60 minutes or with 500 µM H2O2 as a positive control for 15 minutes. Platelets were lysed, and unmodified cysteine thiols were alkylated with 5 µM of 5-IAF; oxidized thiols are unable to be alkylated by 5-IAF. SFK were immunoprecipitated and washed, and 5-IAF was detected by an anti-fluorescein antibody. Total SFK were detected by an anti-Src antibody. (B) oxLDL promotes SFK cysteine sulfenylation over time. (C) SFK cysteine sulfenylation by oxLDL is CD36 dependent. (D) H2O2 is the ROS downstream of CD36 that sulfenylates SFK. Washed human platelets (3 × 108/mL) were pretreated with 1 mM BTD-alkyne (B) in the presence of 1 µg/mL CD36-blocking FA6-152 (FA6) or control IgG antibody (C), or with 1000 U/mL of denatured or functional PEG-catalase (PEG-Cat) (D). The platelets were then stimulated with 50 µg/mL oxLDL up to 60 minutes (B-D), or with 500 µM H2O2 as a positive control for 15 minutes (B). Levels of SFK immunoprecipitated were assessed by UV-dependent stain-free imaging of the Bio-Rad TGX gels. (E) oxLDL/CD36 signaling promotes activation of SFK over time. (F) SFK activation by CD36 is H2O2 dependent. Washed human platelets (3 × 108/mL) were stimulated with 50 µg/mL oxLDL up to 60 minutes or with 500 µM H2O2 as a positive control (E) or with 50 µg/mL oxLDL following pretreatment with 1000 U/mL functional or denatured PEG-catalase (PEG-Cat) (F) for 15 minutes. The cells were lysed with radioimmunoprecipitation assay buffer, and phosphorylated SFK (Y416) were detected by immunoblotting. Total SFK was detected by an anti-Src antibody. P values were determined by 1-way analysis of variance with Dunnett’s posthoc analysis (A-C,E) and by 1-way analysis of variance with Tukey’s post hoc analysis (D,F). N = 3 separate donors (A-F). *P < .05 compared with buffer treatment (A-C,E). *P < .05 with their respective comparisons (D,F). Data are expressed as mean ± SEM.
To determine if SFK sulfenylation was responsible for the decreased 5-IAF alkylation observed upon oxLDL treatment, platelets were pretreated with BTD-alkyne before stimulation with oxLDL. SFK were immunoprecipitated followed by ligation of biotin-PEG3-azide. In inverse correlation to the decreased 5-IAF alkylation, BTD-alkyne incorporation into SFK was increased by H2O2 (Figure 3B). oxLDL stimulation also promoted BTD-alkyne incorporation into SFK, which reached a maximum within 30 to 60 minutes. We examined other potential oxidative cysteine modifications of SFK (ie, protein disulfide formation) (supplemental Figure 3A) and observed no differences upon oxLDL stimulation. We then determined if Fyn or Lyn were sulfenylated upon oxLDL stimulation. Increased sulfenylation of Fyn but not Lyn was observed when platelets were treated with oxLDL (supplemental Figure 3B). These data indicate that platelet SFK are modified by cysteine sulfenylation, consistent with the previously described Src modification by ROS.33
We next determined if SFK sulfenylation by oxLDL was CD36 dependent and found that oxLDL-mediated BTD-alkyne incorporation onto SFK was decreased when platelets were pretreated with a CD36-blocking antibody (FA6) compared with IgG control (Figure 3C). PEG-catalase significantly decreased BTD-alkyne incorporation back to levels in unstimulated platelets, indicating that BTD-alkyne incorporation onto SFK was driven by H2O2 and not by other ROS (Figure 3D). These data suggest that SFK sulfenylation is mediated by H2O2 generated from CD36 signaling.
Mechanistically, SFK oxidation promotes kinase activation, which was probed by blotting for phosphorylated Y416. As a positive control, H2O2 treatment exhibited ∼80% more phosphorylated Y416 relative to unstimulated platelets (Figure 3E), similar to previous findings in a cancer cell line.33 CD36 signaling promoted time-dependent Y416 phosphorylation, which was maximal between 30 and 60 minutes and similar to levels observed with H2O2 treatment. Pretreatment with PEG-catalase abrogated the increase in Src Y416 phosphorylation to background levels (Figure 3F). These data suggest that H2O2 is a signaling effector downstream of CD36 to promote SFK activation.
Labeling cysteine sulfenylation with carbon nucleophiles prevents platelet aggregation by oxLDL/CD36 but not other agonists
Carbon nucleophiles modify sulfenic acids with varying rate constants and differential selectivity toward exposed and buried cysteines.18 1,3-cyclohexanedione (CHD) is the slowest of the nucleophiles tested, with a rate constant of ∼10 M−1s−1, whereas BTD exhibits the fastest rate (∼1700 M−1s−1).18,19 Carbon nucleophiles without an alkyne were used to modify cysteine sulfenic acids (Figure 4A), thereby preventing reversibility of these cysteines back to free thiols or to further oxoforms (supplemental Figure 1). Pretreatment with carbon nucleophiles exhibited varying degrees of inhibition of platelet activation and aggregation by oxLDL (Figure 4B-C). BTD showed the most potent inhibition (50% inhibitory concentration [IC50] = 2.0 ± 0.6 mM) consistent with BTD displaying the fastest reactivity toward sulfenic acids. CHD exhibited the next most potent IC50 (5.9 ± 0.7 mM) despite CHD having the slowest reactivity.18 1-Methylpyrrolidine-2,4-dione and 1-methylpiperidine-2,4-dione both showed weaker IC50 values (9.2 ± 0.8 mM and 10.2 ± 0.5 mM, respectively) relative to BTD and CHD, which may reflect lower selectivity toward modification of sulfenylated cysteines critical for platelet aggregation.18,19 Mechanistically, CD36 signals through SFK and H2O2-mediated activation of ERK5.6 BTD 2 mM prevented oxLDL-induced ERK5 activation (supplemental Figure 3D), suggesting sulfenylation could be an important upstream activator of ERK5.
![Cysteine sulfenylation-selective carbon nucleophiles prevent oxLDL/CD36–mediated platelet aggregation. (A) Structures of the carbon nucleophiles used to selectively target cysteine sulfenylation (Cys-SOH). The red carbon is the site of covalent adduction onto sulfenylated cysteines. (B) Carbon nucleophiles show varying degrees of inhibiting platelet aggregation by oxLDL/CD36. Human platelets (3 × 108/mL) were pretreated with varying concentrations of carbon nucleophiles for 15 minutes prior to stimulation for 15 minutes with 50 µg/mL oxLDL. Platelet aggregation was initiated after 200 µg/mL fibrinogen and 1 mM CaCl2/MgCl2 were added. The percentage of maximum platelet aggregation by oxLDL was plotted and the IC50 calculated from fitted curves using log [inhibitor] vs normalized response nonlinear regression. (C) Representative aggregometry tracing of BTD-mediated inhibition of CD36 signaling. Human platelets (3 × 108/mL) were pretreated with DMSO or up to 5 mM BTD, followed by stimulation with 50 µg/mL oxLDL for 15 minutes. Platelet aggregation was initiated after adding 200 µg/mL fibrinogen and 1 mM CaCl2/MgCl2. BTD does not inhibit platelet activation by physiologic activators ADP and collagen. (D-E) Human platelets (3 × 108/mL) were pretreated with 1 mM BTD for 15 minutes before aggregation induced by the agonists ADP (2 or 20 µM) (D) or collagen-related peptide (CRP; 1 or 10 µg/mL) (E). P values were determined by 1-way analysis of variance with Dunnett’s post hoc analysis (C) and paired Student t test (D-E). **P < .01 compared with no treatment in (C). N ≥ 3 separate donors (B-C), and N = 3 separate donors (D-E). Data are expressed as mean ± SEM. PRD, 1-methylpiperidine-2,4-dione; PYD, 1-methylpyrrolidine-2,4-dione.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/18/10.1182_bloodadvances.2020001609/2/m_advancesadv2020001609f4.png?Expires=1769135943&Signature=1U8n7bzPHOMpHkBeZUc9Mr92EZNx0xckvyrPw68lz08rbYgCdntNO~Cxwdv8AV1LLdMQI9qQBxd7H7v8sykWJlNnxlP22e1dsoh77XRwX~gAtGMGoMrr-HDMF6-5IZphy6NebvEzZrIRgfbeh-Ns18a-jyz7O0zpuBGiANIzYJ4c~qaUhyHR5Kb-nE6LVLGCT4Fh~HPlabNij1-MIsrhYm3enUyApB8yvpawTmtSNlucIcZxi2yFFmvwqcMWSysWY6XYMCGvA2cw1iEs5hj6uHInOCQJyIsHtm4IM5LzMNtlgmH8k9b74szBOfnQkkVlDVwuO6JUAFQHTnfgsMBNPw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Cysteine sulfenylation-selective carbon nucleophiles prevent oxLDL/CD36–mediated platelet aggregation. (A) Structures of the carbon nucleophiles used to selectively target cysteine sulfenylation (Cys-SOH). The red carbon is the site of covalent adduction onto sulfenylated cysteines. (B) Carbon nucleophiles show varying degrees of inhibiting platelet aggregation by oxLDL/CD36. Human platelets (3 × 108/mL) were pretreated with varying concentrations of carbon nucleophiles for 15 minutes prior to stimulation for 15 minutes with 50 µg/mL oxLDL. Platelet aggregation was initiated after 200 µg/mL fibrinogen and 1 mM CaCl2/MgCl2 were added. The percentage of maximum platelet aggregation by oxLDL was plotted and the IC50 calculated from fitted curves using log [inhibitor] vs normalized response nonlinear regression. (C) Representative aggregometry tracing of BTD-mediated inhibition of CD36 signaling. Human platelets (3 × 108/mL) were pretreated with DMSO or up to 5 mM BTD, followed by stimulation with 50 µg/mL oxLDL for 15 minutes. Platelet aggregation was initiated after adding 200 µg/mL fibrinogen and 1 mM CaCl2/MgCl2. BTD does not inhibit platelet activation by physiologic activators ADP and collagen. (D-E) Human platelets (3 × 108/mL) were pretreated with 1 mM BTD for 15 minutes before aggregation induced by the agonists ADP (2 or 20 µM) (D) or collagen-related peptide (CRP; 1 or 10 µg/mL) (E). P values were determined by 1-way analysis of variance with Dunnett’s post hoc analysis (C) and paired Student t test (D-E). **P < .01 compared with no treatment in (C). N ≥ 3 separate donors (B-C), and N = 3 separate donors (D-E). Data are expressed as mean ± SEM. PRD, 1-methylpiperidine-2,4-dione; PYD, 1-methylpyrrolidine-2,4-dione.
Cysteine sulfenylation-selective carbon nucleophiles prevent oxLDL/CD36–mediated platelet aggregation. (A) Structures of the carbon nucleophiles used to selectively target cysteine sulfenylation (Cys-SOH). The red carbon is the site of covalent adduction onto sulfenylated cysteines. (B) Carbon nucleophiles show varying degrees of inhibiting platelet aggregation by oxLDL/CD36. Human platelets (3 × 108/mL) were pretreated with varying concentrations of carbon nucleophiles for 15 minutes prior to stimulation for 15 minutes with 50 µg/mL oxLDL. Platelet aggregation was initiated after 200 µg/mL fibrinogen and 1 mM CaCl2/MgCl2 were added. The percentage of maximum platelet aggregation by oxLDL was plotted and the IC50 calculated from fitted curves using log [inhibitor] vs normalized response nonlinear regression. (C) Representative aggregometry tracing of BTD-mediated inhibition of CD36 signaling. Human platelets (3 × 108/mL) were pretreated with DMSO or up to 5 mM BTD, followed by stimulation with 50 µg/mL oxLDL for 15 minutes. Platelet aggregation was initiated after adding 200 µg/mL fibrinogen and 1 mM CaCl2/MgCl2. BTD does not inhibit platelet activation by physiologic activators ADP and collagen. (D-E) Human platelets (3 × 108/mL) were pretreated with 1 mM BTD for 15 minutes before aggregation induced by the agonists ADP (2 or 20 µM) (D) or collagen-related peptide (CRP; 1 or 10 µg/mL) (E). P values were determined by 1-way analysis of variance with Dunnett’s post hoc analysis (C) and paired Student t test (D-E). **P < .01 compared with no treatment in (C). N ≥ 3 separate donors (B-C), and N = 3 separate donors (D-E). Data are expressed as mean ± SEM. PRD, 1-methylpiperidine-2,4-dione; PYD, 1-methylpyrrolidine-2,4-dione.
We next tested the impact of carbon nucleophiles on platelet activation by “classic” physiologic agonists. Adenosine diphosphate (ADP) activates platelets through its purinergic G protein–coupled receptors, P2Y1/12,36 whereas collagen-related peptide activates platelets through a GPVI pathway independent of G protein–coupled receptors. Treatment of platelets with 1 mM BTD did not affect platelet activation by either low or high concentrations of either agonist (Figure 4D-E). Higher concentrations of BTD (10 mM) showed some inhibition of P2Y1/12 receptor signaling but not the GPVI pathway (supplemental Figure 4A-B), suggesting BTD concentrations <10 mM are required to selectively prevent cysteine modifications by oxLDL. Altogether, cysteine sulfenylation by oxLDL is selective to the CD36 pathway to augment platelet activation and aggregation.
Carbon nucleophiles inhibit procoagulant PSer externalization by oxLDL/CD36 signaling
CD36 promotes procoagulant PSer externalization via cross talk with the collagen-receptor GPVI pathway through apoptotic caspase activation.10 PSer externalization promotes assembly of factor tenase and prothrombinase complexes to generate thrombin, which cleaves soluble fibrinogen to form fibrin.37 Given that redox signaling is required for the cross talk between CD36 and GPVI, we tested the hypothesis that PSer externalization by CD36 signaling requires dynamic cysteine sulfenylation. Flow cytometry quantification of PSer externalization by surface-bound annexin V revealed characteristic time-dependent PSer externalization by oxLDL alone, which reached a maximum between 5 and 15 minutes, with 20% to 25% of the platelet population positive for annexin V binding (Figure 5). oxLDL sensitization before stimulation with the snake venom convulxin (CVX), which activates GPVI similarly to collagen,38 showed augmented PSer externalization with up to 40% to 50% of the population positive for annexin V binding. oxLDL with CVX also showed increased levels of PSer externalization with oxLDL alone or during costimulation with CVX (supplemental Figure 4C). Sulfenic acid modification with either BTD or CHD prevented PSer externalization by oxLDL alone or oxLDL with CVX (Figure 5B-C; supplemental Figure 4C-D), similarly to our previously described observation that PEG-catalase prevented oxLDL-induced PSer externalization.10 These data indicate that cysteine sulfenylation contributes to PSer externalization through the CD36-signaling axis.

Carbon nucleophiles BTD and CHD inhibit oxLDL/CD36–induced PSer externalization. (A) Dot plots of annexin V binding by DMSO or 2 mM BTD treatment in oxLDL stimulation alone or oxLDL pretreatment with CVX stimulation. (B-C) Percent positive annexin V binding in the time course stimulation with oxLDL alone or oxLDL with CVX in BTD- (B) and CHD-treated (C) samples. Washed human platelets (30 ×103/µL) were pretreated with 2 mM BTD or 5 mM CHD for 15 minutes at 37°C. After the addition of 1 mM CaCl2/MgCl2, platelets were stimulated with 50 µg/mL oxLDL for up to 30 minutes. In some conditions, platelets were subjected to 5 minutes of costimulation with 500 ng/mL CVX following the time course treatment with oxLDL. P values were determined by 1-way analysis of variance with 2-stage Benjamin, Krieger, and Yekutieli post hoc analysis. *P < .05 compared with DMSO, no stimulation (no oxLDL, no CVX); #P < .05 compared with DMSO, CVX alone (no oxLDL); ##P < .01 compared with DMSO, CVX alone (no oxLDL); †P < .05 compared with their respective DMSO control; and ††P < .01 compared with their respective DMSO control. N = 6 separate donors for the BTD treatments. N = 3 separate donors for the CHD treatments. Data are expressed as mean ± SEM.
Carbon nucleophiles BTD and CHD inhibit oxLDL/CD36–induced PSer externalization. (A) Dot plots of annexin V binding by DMSO or 2 mM BTD treatment in oxLDL stimulation alone or oxLDL pretreatment with CVX stimulation. (B-C) Percent positive annexin V binding in the time course stimulation with oxLDL alone or oxLDL with CVX in BTD- (B) and CHD-treated (C) samples. Washed human platelets (30 ×103/µL) were pretreated with 2 mM BTD or 5 mM CHD for 15 minutes at 37°C. After the addition of 1 mM CaCl2/MgCl2, platelets were stimulated with 50 µg/mL oxLDL for up to 30 minutes. In some conditions, platelets were subjected to 5 minutes of costimulation with 500 ng/mL CVX following the time course treatment with oxLDL. P values were determined by 1-way analysis of variance with 2-stage Benjamin, Krieger, and Yekutieli post hoc analysis. *P < .05 compared with DMSO, no stimulation (no oxLDL, no CVX); #P < .05 compared with DMSO, CVX alone (no oxLDL); ##P < .01 compared with DMSO, CVX alone (no oxLDL); †P < .05 compared with their respective DMSO control; and ††P < .01 compared with their respective DMSO control. N = 6 separate donors for the BTD treatments. N = 3 separate donors for the CHD treatments. Data are expressed as mean ± SEM.
The carbon nucleophile BTD inhibits enhanced arterial thrombosis in mice fed a high-fat diet
Platelet reactivity in dyslipidemia manifests as a prothrombotic phenotype in mice upon vascular injury. Using the FeCl3-induced carotid artery injury model, the absence of CD36 or its downstream signaling molecules (eg, MAPK and ERK5) did not affect thrombosis in normolipidemic conditions.2,9,10,25,39 However, mice fed an atherogenic high-fat, high-cholesterol diet displayed accelerated vessel occlusion after injury compared with chow-fed animals. This thrombotic diathesis was prevented when CD36 or any of its downstream signaling effectors were absent or inhibited in platelets.2
We next tested the hypothesis that cysteine sulfenylation contributes to dyslipidemia-induced enhanced arterial thrombosis in vivo. Mice on a high-fat diet displayed accelerated vessel occlusion at a median of 6.5 minutes compared with the median occlusion time of 11 minutes in chow-fed mice (Figure 6A-B; supplemental Videos 1-2). BTD dose dependently attenuated the vessel occlusion time in mice fed a high-fat diet (supplemental Videos 3-5), with BTD 25 mg/kg treatment showing vessel occlusion time comparable to that of vehicle-treated, chow-fed mice (median of 12 minutes and 11 minutes, respectively). Kaplan-Meyer analysis (Figure 6C) revealed that the frequency of total vessels remaining open in mice treated with BTD 25 mg/kg and fed a high-fat diet was similar to that of vehicle-treated, chow-fed mice.
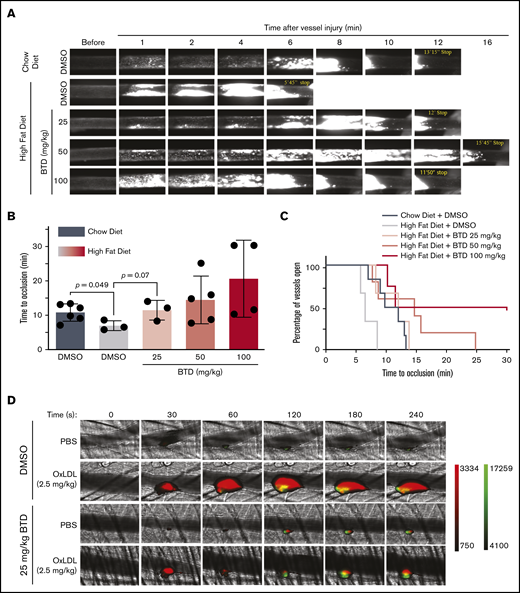
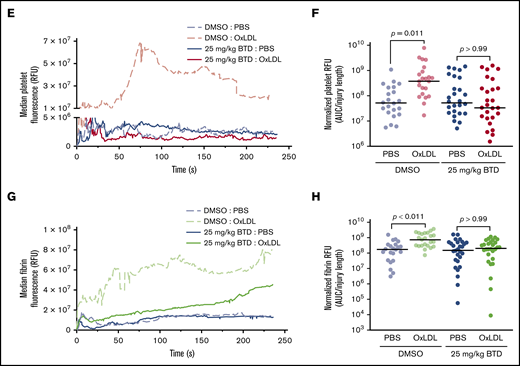
BTD carbon nucleophile decreases arterial thrombosis in dyslipidemia. BTD inhibits the prothrombotic platelet phenotype in “Western” high-fat diet-fed mice to chow-fed control levels using the FeCl3-induced carotid artery thrombosis model. (A) Intravital microscopy video images of the carotid artery from mice fed chow or a high-fat diet treated with vehicle control or 25 mg/kg BTD. (B-C) Histograms of time to occlusion (B), and Kaplan-Meier curves of the fraction of vessels not occluded (C) are shown (P = .01 between Chow Diet + DMSO vs High Fat Diet + DMSO; P = .46 between Chow Diet + DMSO vs High Fat Diet + 25 mg/kg BTD). BTD rescued the enhanced platelet and fibrin accumulation induced by oxLDL in the laser ablation cremasteric artery thrombosis model (C). (D) Intravital microscopy images of the cremasteric artery from intravenously injected PBS or 2.5 mg/kg oxLDL-injected mice treated with vehicle or 25 mg/kg BTD. Platelets are red and fibrin is green. (E,G) Median integrated fluorescence intensity over time of platelets (E) and fibrin (G) are shown. (F,H) Quantification of the normalized platelet (F) and fibrin (H) accumulation as area under the curve (AUC) by the length of the injury are presented. In both thrombosis models, DMSO (as the vehicle treatment) and BTD were prepared in 20% v/v cremophore in PBS and were injected intraperitoneally 3 hours before injury. P values were determined by unpaired Student t test (B), log-rank (Mantel-Cox) Kaplan-Meier tests (C), and 1-way analysis of variance with Kruskal-Wallis pairwise comparisons (F,H). For the FeCl3 thrombosis model, N = 6 mice for vehicle control treatment in conditions of a chow diet. N = 3 mice for vehicle control treatment, 3 mice for 25 mg/kg BTD treatment, 5 mice for 50 mg/kg BTD treatment, and 4 mice for 100 mg/kg BTD treatment in conditions of a high-fat diet. For the laser ablation thrombosis model, N = 3 mice for DMSO and 3 mice for BTD treatment and ≥22 injuries per PBS or oxLDL cohort of thrombosis. Data are expressed as mean ± SD.
BTD carbon nucleophile decreases arterial thrombosis in dyslipidemia. BTD inhibits the prothrombotic platelet phenotype in “Western” high-fat diet-fed mice to chow-fed control levels using the FeCl3-induced carotid artery thrombosis model. (A) Intravital microscopy video images of the carotid artery from mice fed chow or a high-fat diet treated with vehicle control or 25 mg/kg BTD. (B-C) Histograms of time to occlusion (B), and Kaplan-Meier curves of the fraction of vessels not occluded (C) are shown (P = .01 between Chow Diet + DMSO vs High Fat Diet + DMSO; P = .46 between Chow Diet + DMSO vs High Fat Diet + 25 mg/kg BTD). BTD rescued the enhanced platelet and fibrin accumulation induced by oxLDL in the laser ablation cremasteric artery thrombosis model (C). (D) Intravital microscopy images of the cremasteric artery from intravenously injected PBS or 2.5 mg/kg oxLDL-injected mice treated with vehicle or 25 mg/kg BTD. Platelets are red and fibrin is green. (E,G) Median integrated fluorescence intensity over time of platelets (E) and fibrin (G) are shown. (F,H) Quantification of the normalized platelet (F) and fibrin (H) accumulation as area under the curve (AUC) by the length of the injury are presented. In both thrombosis models, DMSO (as the vehicle treatment) and BTD were prepared in 20% v/v cremophore in PBS and were injected intraperitoneally 3 hours before injury. P values were determined by unpaired Student t test (B), log-rank (Mantel-Cox) Kaplan-Meier tests (C), and 1-way analysis of variance with Kruskal-Wallis pairwise comparisons (F,H). For the FeCl3 thrombosis model, N = 6 mice for vehicle control treatment in conditions of a chow diet. N = 3 mice for vehicle control treatment, 3 mice for 25 mg/kg BTD treatment, 5 mice for 50 mg/kg BTD treatment, and 4 mice for 100 mg/kg BTD treatment in conditions of a high-fat diet. For the laser ablation thrombosis model, N = 3 mice for DMSO and 3 mice for BTD treatment and ≥22 injuries per PBS or oxLDL cohort of thrombosis. Data are expressed as mean ± SD.
Using the laser ablation cremaster artery thrombosis model with intravenous oxLDL injection as a surrogate for dyslipidemia,25,26 we found that oxLDL increased both platelet and fibrin accumulation over time (Figure 6D-H; supplemental Videos 6-7) compared with PBS injection. Native unoxidized LDL did not increase platelet and fibrin accumulation (supplemental Figure 5A-E; supplemental Videos 8-9). Laser ablation injury sizes40 were similar between all conditions (supplemental Figure 5F-G). BTD 25 mg/kg pretreatment rescued both enhanced platelet and fibrin accumulation by oxLDL to levels observed with PBS control conditions (supplemental Videos 10-11). These data strongly suggest that cysteine sulfenylation in dyslipidemia promotes arterial thrombosis and that chemoselective modification of sulfenylation with carbon nucleophiles decreases the prothrombotic phenotype.
Discussion
Platelet activation in dyslipidemia increases risk for clinically significant thrombotic events. The signaling mechanisms that promote platelet activation in these settings are incompletely defined and are mediated in part by CD36 redox signaling. As modeled in Figure 7, we report that H2O2 accumulates upon CD36 recognition of its model ligand oxLDL. This accumulation results in oxidative cysteine modification important for platelet activation. In particular, we show that SFK are sulfenylated, which promotes CD36 signaling. oxLDL/CD36–mediated platelet activation and aggregation were inhibited using carbon nucleophiles that modify sulfenic acids important in mediating proaggregatory functions. Sulfenylation is also important for cross talk between the CD36 and GPVI receptors leading to PSer externalization, recruitment of procoagulant factors tenase and prothrombinase, and fibrin deposition. Sulfenylation is therefore a selective oxidative modification to CD36 signaling and a potential target to prevent arterial thrombosis in dyslipidemia.
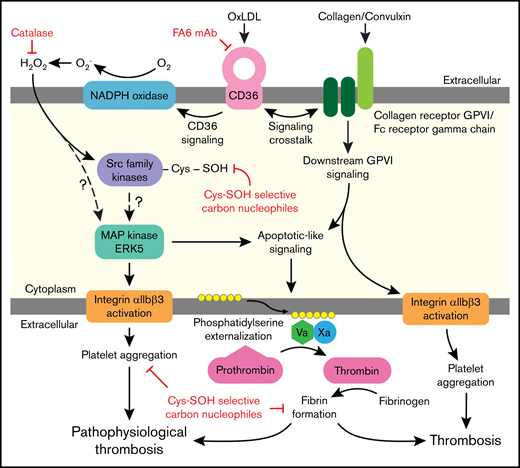
Illustration of platelet CD36 redox signaling in arterial thrombosis. Oxidized lipids are recognized by scavenger receptor CD36 on the platelet surface. CD36 signal transduction leads to H2O2 generation from NADPH oxidase. H2O2 promotes oxidative cysteine modification of cellular regulators of platelet activation, including SFK. Specifically, SFK cysteines undergo sulfenylation, which is a potential mechanism for activation of redox-sensitive MAPK ERK5. ERK5 links platelet CD36 signaling to 2 functional phenotypes: (1) platelet aggregation mediated by integrin αIIbβ3; and (2) procoagulant PSer externalization required for assembly of the prothrombinase complex to form fibrin from activated thrombin. The CD36 signaling pathway also cross talks with the collagen receptor GPVI pathway to augment PSer externalization. This redox-regulated CD36 signaling pathway promotes pathophysiologic thrombosis in dyslipidemia. In the absence of CD36 signaling, collagen-mediated GPVI activation promotes platelet integrin αIIbβ3 activation and procoagulant PSer externalization during thrombosis.
Illustration of platelet CD36 redox signaling in arterial thrombosis. Oxidized lipids are recognized by scavenger receptor CD36 on the platelet surface. CD36 signal transduction leads to H2O2 generation from NADPH oxidase. H2O2 promotes oxidative cysteine modification of cellular regulators of platelet activation, including SFK. Specifically, SFK cysteines undergo sulfenylation, which is a potential mechanism for activation of redox-sensitive MAPK ERK5. ERK5 links platelet CD36 signaling to 2 functional phenotypes: (1) platelet aggregation mediated by integrin αIIbβ3; and (2) procoagulant PSer externalization required for assembly of the prothrombinase complex to form fibrin from activated thrombin. The CD36 signaling pathway also cross talks with the collagen receptor GPVI pathway to augment PSer externalization. This redox-regulated CD36 signaling pathway promotes pathophysiologic thrombosis in dyslipidemia. In the absence of CD36 signaling, collagen-mediated GPVI activation promotes platelet integrin αIIbβ3 activation and procoagulant PSer externalization during thrombosis.
Until recently, measuring specific ROS in platelets has been challenging due to a lack of selective probes. Boronate-based probes are selective for H2O2 and peroxynitrite with second order rate constants of ∼1.5 and ∼1 × 106 M−1s−1, respectively.21,27 Our study showed that H2O2 was generated by CD36 signaling in platelets (Figure 1). The rate constants should be considered when interpreting the data because the slow reactivity of boronates, despite their selectivity toward peroxides, will significantly lag behind relevant cellular signaling and mechanisms of peroxide degradation. Although PEG-catalase allowed determination that H2O2 was primarily responsible for CBA oxidation to COH, we do not exclude the possibility of peroxynitrite generation. Because peroxynitrite can also oxidize cysteines to sulfenic acids, the net result on downstream CD36 signaling would likely be similar to that of H2O2.41 Importantly, COH fluorescence in platelets by oxLDL was inhibited by the CD36-blocking antibody and by addition of PEG-catalase (Figure 1F-G). These data suggest a CD36- and cellular-dependent H2O2 mechanism.
Sites of cysteine sulfenylation can selectively react with carbon nucleophiles.18 Using an alkyne-containing benzothiazine-based probe (BTD-alkyne), we report sulfenylation in platelets (Figure 2). The apparent kinetics of cysteine sulfenylation by CD36 signaling as detected by BTD should not be directly compared with the kinetics of peroxide generation as detected by boronates because BTD reacts with sulfenic acids with a second order rate constant of 1700 M−1s−1, much faster than the reaction of boronates with H2O2.27 We also tested for other cysteine modifications (eg, disulfide formation) (supplemental Figure 3A); however, we observed no significant changes in these modifications upon oxLDL stimulation. We do not exclude a role for further cysteine oxoforms in CD36 signaling (supplemental Figure 1). We also observed cysteine sulfenylation through thrombin-mediated activation of protease-activated receptor 1/4 (supplemental Figure 3E) but minimal to no increased sulfenylation with ADP and CVX (supplemental Figure 3F-G). Sulfenylation by classic agonists compared with oxLDL require further investigation because proteins in close proximity to the source of H2O2 are affected more than distal proteins.42,43
SFK are direct mediators of both platelet activation and inhibitory signaling.44 Human platelets express several SFK members, including Src, Fyn, and Lyn.44 Cys277 on c-Src (Cys280 in human Src) was shown to be sulfenylated and important for the activity of the protein.33 We showed that SFK cysteines were sulfenylated by a CD36- and H2O2-dependent mechanism (Figure 3). Furthermore, Fyn and Lyn are specific members recruited to CD36 upon oxLDL binding.5,32 Fyn, but not Lyn, was sulfenylated in response to oxLDL (supplemental Figure 3B). The alternatively spliced Fyn isoform 2 is expressed in hematopoietic lineages compared with brain dominant isoform 1.45 Cys278 in Fyn isoform 2 is homologous to Cys280 of human Src (supplemental Figure 3C); neither Lyn isoforms nor other Fyn isoforms have this cysteine. These data are consistent with our previous report that Fyn is the SFK member preferentially activated by platelet CD36 signaling.5
To test the functional impact of sulfenylation, we used several carbon nucleophiles that modify sites of cysteine sulfenylation and found sulfenylation is important for platelet proaggregatory and procoagulant phenotypes induced by the oxLDL/CD36 pathway (Figures 4 and 5). The degree of inhibition by these nucleophiles could be related to their specificity toward distinct cysteines.18 Lower BTD concentrations (1 and 2 mM) did not prevent platelet aggregation induced by ADP and collagen; higher BTD concentrations (10 mM) induced nonselective interactions as indicated by inhibiting the P2Y1/12 pathway (supplemental Figure 4A) but not the GPVI pathway (supplemental Figure 4B). The nucleophiles also decreased procoagulant PSer externalization by oxLDL alone and oxLDL with CVX. PSer externalization by CVX alone (no oxLDL) was decreased to some extent by the carbon nucleophiles, which suggests a preferential role for sulfenylation in procoagulant functions by classic activators. This mechanism may be related to the specific SFK involved, as we recently proposed that SFK link the 2 pathways.10,39
Dyslipidemia is associated with a chronic state of oxidative stress and induces a prothrombotic phenotype in platelets through CD36.2,6,9,25 Using a high-fat, high-cholesterol diet10,25,46 and intravenous injection of oxLDL25,26 as models of dyslipidemia, we found that dyslipidemic conditions promoted a prothrombotic phenotype in vivo in the FeCl3 and laser ablation–induced arterial thrombosis models (Figure 6). Importantly, chemoselective modification of cysteine sulfenylation with BTD rescued the prothrombotic and procoagulant nature of dyslipidemia to that of chow-dieted or intravenous PBS injection controls, indicating an important role for cysteine sulfenic acids in promoting thrombosis in oxidant stress conditions. Titrating the probe to higher concentration than 25 mg/kg may delay vascular occlusion times, consistent with supplemental Figure 4A that higher concentration of the probe enhances nonselective interaction with cellular pathways.
In summary, our studies identified cysteine sulfenylation as an oxidative posttranslational modification important in CD36 signaling that could be exploited to prevent platelet activation in conditions associated with oxidant stress, including dyslipidemia, diabetes mellitus, and chronic inflammation.47
Requests regarding data and protocol may be submitted to brismith@mcw.edu, rsilverstein@mcw.edu, or rflaumen@bidmc.harvard.edu.
Acknowledgments
The authors thank members of the Redox Biology Program and the Free Radical Research Center of the Medical College of Wisconsin for insightful discussions.
This work was supported by National Institutes of Health (NIH), National Heart, Lung, and Blood Institute grants R01HL111614 (R.L.S), R01HL142152 (R.L.S.), R01HL111614-S (M.Y. as trainee), T32HL134643 and T32HL007917 (M.Y. as trainee), R35HL135775 (R.F.), and R15HL145573 (W.L.), and NIH, National Institute of General Medical Sciences grants R35GM128840 (B.C.S.) and R01GM102187 (K.S.C.). This work was also supported by American Heart Association grant 15SDG25830057 (B.C.S.) and the Medical College of Wisconsin Cardiovascular Center’s A. O. Smith Fellowship Scholars Program (M.Y. as trainee).
Authorship
Contribution: M.Y. designed and performed the experiments and wrote the manuscript; W.L. designed and performed the in vivo FeCl3 thrombosis experiments, and edited and wrote parts of the manuscript; C.H. performed HPLC experiments and parts of the click chemistry experiments; W.C. performed platelet aggregometry experiments; H.Y. assisted with the in vivo FeCl3 thrombosis experiments; S.L.W.-S. provided a protocol for click chemistry, provided input on cysteine sulfenylation data, and edited the manuscript; K.S.C. and R.B.F. provided BTD, input on data relating to sulfenic acid–selective carbon nucleophiles, and edited the manuscript; J.Z. provided CBA and COH and input on H2O2 measurements; and R.L.S., R.F., and B.C.S. supervised the project and edited the manuscript. All the authors analyzed the data.
Conflict-of-interest disclosure: R.F. is a founder and consultant for Platelet Diagnostics. The remaining authors declare no competing financial interests.
Correspondence: Brian C. Smith, Medical College of Wisconsin, Department of Biochemistry, Room 356, 8701 West Watertown Plank Rd, Milwaukee, WI 53226; e-mail: brismith@mcw.edu; or Roy L. Silverstein, Medical College of Wisconsin, Hub for Medical Collaboration, Room 8745, 8701 West Watertown Plank Rd, Milwaukee, WI 53226; e-mail: rsilverstein@mcw.edu.

