

Key Points
AMG 701 potently induced autologous RRMM patient cell lysis, T-cell proliferation, and differentiation leading to higher CD8/CD4 ratios.
Lenalidomide and pomalidomide further enhanced T-cell modulation, acting synergistically with AMG 701 to prevent myeloma relapse in vivo.

Abstract
We investigated here the novel immunomodulation and anti–multiple myeloma (MM) function of T cells engaged by the bispecific T-cell engager molecule AMG 701, and further examined the impact of AMG 701 in combination with immunomodulatory drugs (IMiDs; lenalidomide and pomalidomide). AMG 701 potently induced T-cell–dependent cellular cytotoxicity (TDCC) against MM cells expressing B-cell maturation antigen, including autologous cells from patients with relapsed and refractory MM (RRMM) (half maximal effective concentration, <46.6 pM). Besides inducing T-cell proliferation and cytolytic activity, AMG 701 also promoted differentiation of patient T cells to central memory, effector memory, and stem cell–like memory (scm) phenotypes, more so in CD8 vs CD4 T subsets, resulting in increased CD8/CD4 ratios in 7-day ex vivo cocultures. IMiDs and AMG 701 synergistically induced TDCC against MM cell lines and autologous RRMM patient cells, even in the presence of immunosuppressive bone marrow stromal cells or osteoclasts. IMiDs further upregulated AMG 701–induced patient T-cell differentiation toward memory phenotypes, associated with increased CD8/CD4 ratios, increased Tscm, and decreased interleukin 10–positive T and T regulatory cells (CD25highFOXP3high), which may downregulate T effector cells. Importantly, the combination of AMG 701 with lenalidomide induced sustained inhibition of MM cell growth in SCID mice reconstituted with human T cells; tumor regrowth was eventually observed in cohorts treated with either agent alone (P < .001). These results strongly support AMG 701 clinical studies as monotherapy in patients with RRMM (NCT03287908) and the combination with IMiDs to improve patient outcomes in MM.
Introduction
Immunotherapy targeting tumor cells and their surrounding immune microenvironment has significantly improved patient outcomes in multiple myeloma (MM).1,2 Specifically, monoclonal antibodies targeting CD38 (daratumumab, isatuximab) or SLAMF7 (elotuzumab) induce significant immune effector cell–mediated killing of MM cells, and can partially restore function of immune effector cells including T and natural killer (NK) cells.2-8 However, relapse of disease is common and patients with relapsed or refractory MM (RRMM) have a poor prognosis.9 An increased number of prior lines of anti-MM treatments is associated with increased evolution, survival, and proliferation of drug-resistant clones, ultimately leading to drug-resistant minimal residual disease (MRD), clinical relapse, and treatment failure.10-12 Thus, there remains an urgent need to develop novel immunotherapeutic strategies with different mechanisms of action and greater potency to overcome drug resistance and eliminate disease in patients with RRMM.
The conventional bispecific T-cell engager (BiTE) immuno-oncology therapies are composed of 2 binding domains targeting CD3 on T cells and tumor-associated antigens on tumor cells.13-15 Upon concurrent binding of target cells and T cells, BiTE molecules induce T-cell activation and cytokine production, and formation of cytolytic immunological synapses, leading to T-cell–dependent cellular cytotoxicity (TDCC) of target cells.13 BiTE molecules elicit cytolysis without requiring antigen-presenting cells, a major histocompatibility complex class I/peptide complex, or costimulatory molecules. The first BiTE molecule targeting MM cells, AMG 420 (formerly BI 836909), is directed against B-cell maturation antigen (BCMA),16 a tumor antigen selectively and highly expressed on patient MM cells.17-22 AMG 420 has demonstrated the first proof of concept for anti-BCMA BiTE therapy with impressive clinical activity: a 70% overall response rate at 400 μg per day, including some complete responses with MRD negativity for up to >1 year in heavily pretreated RRMM patients in a first-in-human phase 1 clinical trial.23 Such clinical responses are comparable to responses seen with chimeric antigen receptor (CAR) BCMA T cells in RRMM, which require T-cell engineering and ex vivo expansion followed by adoptive transfer into patients.24-26 Like the other conventional BiTE molecules, including blinatumomab, which is approved for the treatment of RR CD19+ acute lymphoblastic leukemia and other B hematological malignancies,16,27,28 AMG 420 has a short half-life, making continuous infusion a necessary therapeutic approach to achieve adequate drug levels.15,23,29,30 One approach to enhancing convenience for patients is to generate BiTE molecules with longer serum half-life to enable once-weekly dosing.
Lenalidomide (len) and pomalidomide (pom), 2 immunomodulatory drugs (IMiDs), are routinely used as a therapeutic backbone or partners in various MM treatment combinations throughout all disease settings.31-35 IMiDs have direct antiproliferative and proapoptotic effects on MM, in addition to indirect anti-MM activity through immunomodulation of multiple immune effector cells, which decrease the immunosuppressive effects of various bone marrow (BM) nonmyeloma accessory cells. IMiDs costimulate immune effector cells (T, NK) and block immunosuppressive BM accessory cells, that is, myeloid suppressor cells, regulatory T (Treg) and regulatory B (Breg) cells, via cell-cell contact and cytokine-mediated mechanisms.4,36-42 In recent preclinical studies, IMiDs coupled with CAR T cells had increased anticancer activity compared with CAR T cells alone,43-45 suggesting that the combination of IMiDs with other novel immunotherapeutic modalities may show enhanced antitumor activity. Until now, the combination of IMiDs with BiTE molecule treatment has not been investigated in MM.
Herein, we study the therapeutic potency of, and immunomodulation by, AMG 701, a serum half-life extended anti-BCMA BiTE molecule, in ex vivo coculture and in vivo preclinical human MM models. We further investigate promising combination treatments with IMiDs and AMG 701 to further augment anti-MM immunity in vitro and in vivo.
Materials and methods
Reagents
AMG 701 was constructed by recombinant DNA technology and produced in supernatants from stably transfected CHO cells and purified by chromatography.46 All other reagents for various assays are detailed in supplemental Methods.
FC-based analysis
Quantitative flow cytometry (FC)–based analysis (supplemental Methods) was used for BCMA expression quantification, TDCC assays under monotreatment or combination treatment settings with len or pom, apoptotic studies, degranulation of effector cells, intracellular cytokine expression, various T-cell phenotypes, and memory cell differentiation.
NCI-H929 (H929) xenograft model of human MM
NCI-H929 (H929) cells (5 × 106) were injected subcutaneously into the right dorsal flank of sublethally irradiated (2 Gy) female SCID mice on day 1. Human pan-T cells expanded in vitro (5 × 106; T-cell isolation, activation/expansion kits from Miltenyi Biotec) were injected intraperitoneally on day 12 into preallocated animals. The animals were randomized into treatment groups on day 15 based on tumor size (group mean tumor volume ∼260 mm3) and treatment with AMG 701, len, or both was initiated on the same day (supplemental Figure 7A). AMG 701 (0.25 mg/kg per administration) was administered by lateral tail vein once weekly starting on day 15 for 5 weeks. len (0.2 mg/kg per administration) was administered intraperitoneally once daily from days 15 to 46. Tumor growth was monitored by external caliper measurements, and tumor volumes were calculated) sing a standard hemiellipsoid formula. Values represent mean tumor sizes (in mm3) ± standard error of the mean (SEM). The study was terminated at day 47. Mice were euthanized when tumor diameter exceeded 15 mm in size. All animal studies were conducted under approved institutional animal care and use committee protocols in Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)–accredited facilities and in accordance with the German Animal Welfare Law with permission from the local authorities and within the guidelines of AAALAC international standards.
Statistics
All statistical analysis was performed using GraphPad Prism (v8; GraphPad Software, La Jolla, CA). P < .05 was considered statistically significant, determined using 1-way analysis of variance (ANOVA), with Bonferroni post hoc comparison (for >3 groups) or a 2-tailed unpaired Student t test (for 2 groups). In vivo data were analyzed using 1-way ANOVA with a Tukey multiple comparison test. Ex vivo data were analyzed using the Wilcoxon matched-pairs signed rank test or Student t test. Evaluation of combination index (CI) for drug interactions was performed using the Chou-Talalay method.47
Results
AMG 701 induced TDCC against MM cells including autologous patient cells resistant to current anti-MM therapies
AMG 701 potently induced TDCC against MM cell lines expressing various BCMA protein levels18,48 when cocultured for 24 hours with healthy donor T cells (n = 5) at an effector-to-target (E:T) ratio of 10:1 (Figure 1A; supplemental Figure 1A-C). Using FC analysis to determine percent lysis of CD138+ MM cells (supplemental Methods), the 50% effective concentration (EC50) values for AMG 701 ranged from 0.85 ± 0.64 pM to 28.03 ± 13.68 pM (n = 8 MM cell lines; supplemental Figure 1C). Furthermore, results from FC-based apoptosis analysis showed >80% to 92% annexin V+CD138+ MM cells following 1- day coculture of IMiD-resistant MM cell lines with AMG 701 (93.4 pM; 10 ng/mL) and patient T cells (n = 3) (Figure 1B; supplemental Figure 1D). MM1S(R) and H929(R) cells are significantly resistant to both len and pom, compared with their parent cell lines.22,48

AMG 701 induced potent T-cell–redirected lysis of MM cell lines and autologous MM patient cells from RRMM. (A-B) MM cell lines sensitive and resistant to current MM therapies were incubated with healthy donor T cells (n = 5) at an E:T ratio of 10 to 1 (10:1) in the presence of AMG 701 for 24 hours, followed by quantitative FC analysis to determine percent lysis of CD138+ MM cells (A) and percentage of apoptotic/dead IMiD-resistant MM cells (B). MM1S(R) and H929(R), resistant to both len and pom, are derived from MM1S and H929, respectively.48 Data represent the means (percent lysis) ± standard deviations (SDs) of 3 to 5 independent experiments using T effector cells from healthy donors (n = 5) (A) and patients (n = 3) (B), each performed in triplicate at each dose. Error bars indicate SDs. Also shown in B (left panel) are representative dot plots of the percentage of apoptotic/dead in CD138+ cells from IMiD-resistant MM cell lines. (C-D) AMG 701 was added into mixtures of patient T effector cells (n = 3) and the same cell number of MM1S target cells (E:T=1:1) for 6 hours, followed by FC analysis to determine the percentage of cells expressing surface CD107a (degranulation) (C) and the percentage of Th1 cytokines (D) in CD4 and CD8 T subsets. (E) BMMCs purified from BM aspirates of RRMM patients (n = 10) were directly incubated with AMG 701 for 24 hours in triplicate at each concentration, followed by FC analysis to measure the percent lysis of CD138+ patient MM cells. All data are shown as means ± SDs. **P < .005; ***P < .001; ****P < .0001.
AMG 701 induced potent T-cell–redirected lysis of MM cell lines and autologous MM patient cells from RRMM. (A-B) MM cell lines sensitive and resistant to current MM therapies were incubated with healthy donor T cells (n = 5) at an E:T ratio of 10 to 1 (10:1) in the presence of AMG 701 for 24 hours, followed by quantitative FC analysis to determine percent lysis of CD138+ MM cells (A) and percentage of apoptotic/dead IMiD-resistant MM cells (B). MM1S(R) and H929(R), resistant to both len and pom, are derived from MM1S and H929, respectively.48 Data represent the means (percent lysis) ± standard deviations (SDs) of 3 to 5 independent experiments using T effector cells from healthy donors (n = 5) (A) and patients (n = 3) (B), each performed in triplicate at each dose. Error bars indicate SDs. Also shown in B (left panel) are representative dot plots of the percentage of apoptotic/dead in CD138+ cells from IMiD-resistant MM cell lines. (C-D) AMG 701 was added into mixtures of patient T effector cells (n = 3) and the same cell number of MM1S target cells (E:T=1:1) for 6 hours, followed by FC analysis to determine the percentage of cells expressing surface CD107a (degranulation) (C) and the percentage of Th1 cytokines (D) in CD4 and CD8 T subsets. (E) BMMCs purified from BM aspirates of RRMM patients (n = 10) were directly incubated with AMG 701 for 24 hours in triplicate at each concentration, followed by FC analysis to measure the percent lysis of CD138+ patient MM cells. All data are shown as means ± SDs. **P < .005; ***P < .001; ****P < .0001.
Following 6-hour coincubation with MM1S target cells, increasing concentrations of AMG 701 increased the percentages of patient T cells (n = 3) that were positive for surface CD107a expression and T helper 1 (Th1) cytokines (Figure 1C-D). In contrast, neither CD107a nor cytokine expression was induced in patient T cells cultured with control medium only or cultured in the absence of MM target cells (data not shown).
Importantly, using BM mononuclear cells (BMMCs) from RRMM patients (n = 10) without adding peripheral blood mononuclear cells (PBMCs) from the same individual, treatment with AMG 701 for 24 hours induced autologous patient CD138+ RRMM cell lysis in a dose-dependent manner, with EC50 values ranging from 6.89 to 46.57 pM. (Figure 1E; supplemental Figure 1E). Patient BMMC sample E:T ratios varied from ∼20:1 to 3:1 and contained non-MM accessory cells. Despite incomplete killing in some samples due to patient T-cell dysfunction or exhaustion, these data indicate that AMG 701 can consistently induce autologous RRMM patient cell lysis.
AMG 701 potently induced T-cell proliferation associated with transient expression of immune-checkpoint and costimulatory molecules, more prominently in CD8 vs CD4 subsets
Next, in cocultures of CellTrace violet (CTV)–labeled patient T cells with RPMI8226 MM cells, AMG 701 (93 pM) induced patient T-cell proliferation at day 4 in a dose-dependent manner. Greater proliferation was observed in CD8 vs CD4 T-cell subsets (Figure 2A left panels). This proliferation was associated with robust induction of the T-cell activation markers CD25 and CD69 (Figure 2A right panel). This effect was further evaluated with additional patient T cells (n = 3) cocultured with other MM target cell lines (n = 3; Figure 2B). AMG 701 induced consistently greater proliferation of CD8 than CD4 cells at a lower dose (9.3 pM; P < .05), but the effect was less consistent at a higher dose (93.4 pM) because almost all MM cells were depleted by day 4. The relative expression of key immune-checkpoint proteins (PD1, TIM3, LAG3) and costimulation molecules (CD28, 4-1BB) induced by AMG 701 was examined on T cells from 7 to 10 healthy donors cocultured with MM cells using mean fluorescence intensity (MFI) ratios normalized to the MFI values of control groups on day 0. Time-course studies showed a transient induction in these proteins, with significantly (Wilcoxon matched-pairs signed-rank test, P < .05) increased levels at day 4 followed by decreased expression at day 7, when compared with control groups (supplemental Figure 2A-B). A transient, but not persistent, upregulation of these checkpoint markers in AMG 701–treated T cells cocultured with MM cells suggests activation, rather than exhaustion, of T cells.49
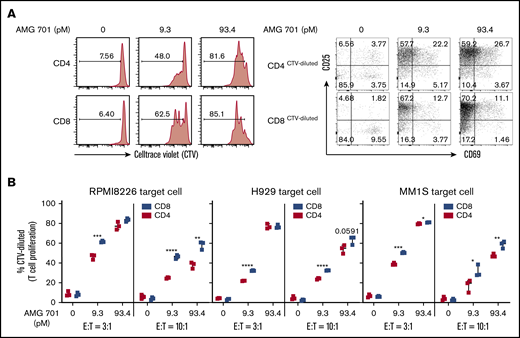
AMG 701 specifically induced proliferation of patient T cells cocultured with MM cells, more prominently in CD8 vs CD4 subsets. (A) Purified patient T cells were labeled with CTV and cocultured with RPMI8226 MM target cells (E:T = 3:1) in the presence of AMG 701 for 4 days, followed by quantitative FC analysis to evaluate the percentage of proliferative (CTV-diluted or CTV−) CD4 and CD8 T subsets (left panels). CD25 and CD69, as late and early activation markers, respectively, were also examined in the proliferative T-cell subsets (right panels). (B) Indicated MM target cell lines (n = 3) were coincubated with CTV-labeled patient T cells (n = 3) at 2 indicated E:T ratios in the presence of AMG 701 (0, 9.3, 93.4 pM). Data are shown as the percentage of CTV-diluted (means ± SDs) in CD4 and CD8 T subsets. Three independent experiments were done with T cells from 3 patients. Each experiment was performed in triplicate at each condition. *P < .05; **P < .005; ***P < .001; ****P < .0001.
AMG 701 specifically induced proliferation of patient T cells cocultured with MM cells, more prominently in CD8 vs CD4 subsets. (A) Purified patient T cells were labeled with CTV and cocultured with RPMI8226 MM target cells (E:T = 3:1) in the presence of AMG 701 for 4 days, followed by quantitative FC analysis to evaluate the percentage of proliferative (CTV-diluted or CTV−) CD4 and CD8 T subsets (left panels). CD25 and CD69, as late and early activation markers, respectively, were also examined in the proliferative T-cell subsets (right panels). (B) Indicated MM target cell lines (n = 3) were coincubated with CTV-labeled patient T cells (n = 3) at 2 indicated E:T ratios in the presence of AMG 701 (0, 9.3, 93.4 pM). Data are shown as the percentage of CTV-diluted (means ± SDs) in CD4 and CD8 T subsets. Three independent experiments were done with T cells from 3 patients. Each experiment was performed in triplicate at each condition. *P < .05; **P < .005; ***P < .001; ****P < .0001.
AMG 701 promoted T-cell differentiation including stem cell–like memory phenotype following coculture with MM cell lines
To study whether AMG 701 induced differentiation of T cells into memory phenotypes following coculture with MM cell lines ex vivo, CD45RA and CD62L expression was examined. In cocultures with MM1S target cells, AMG 701 (93 pM), but not the control BiTE molecule, significantly (P < .05) induced differentiation of CD4 and CD8 T subsets (13 healthy donors) from naive T cells (CD45RA+CD62L+) toward central memory (CM; CD45RA−CD62L+) and effector memory (EM; CD45RA−CD62L−) T cells at day 4 and even more so by day 7 (Figure 3A; supplemental Figure 3A,C). Compared with control groups, AMG 701 significantly increased the percentage of CM and EM cells in CD4 (P < .05) and CD8 (P < .005) T cells (Figure 3A left). During this period, the ratios of CD8/CD4 T cells were significantly increased in AMG 701–treated vs control groups (P < .05 at day 4; P < .0001 at day 7; Figure 3A right). Significantly increased percentages of CM plus EM cells in CD4 and CD8 T-cell subsets were also seen when T cells from patients (n = 3) were cocultured with H929 MM cells (P < .005; Figure 3B; supplemental Figure 3B-C) or RPMI8226 target MM cells (P < .001, Figure 3; supplemental Figure 3C), starting at day 4 and increasing to day 7. CD8/CD4 ratios also continued to increase from day 4 to day 7 (P < .005).
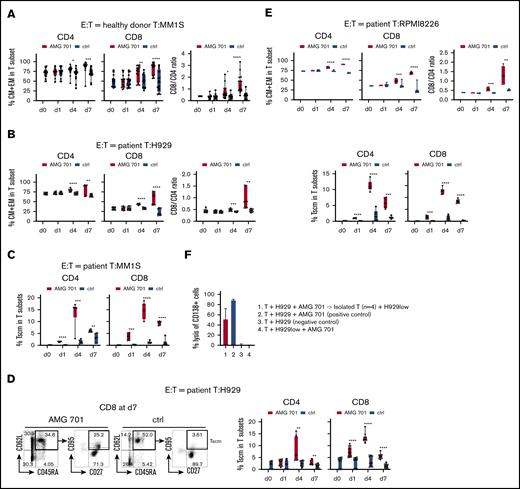
AMG 701 induced T-cell differentiation including scm phenotype, associated with further increases in CD8/CD4 ratios. T cells from donors (A; n = 13) or MM patients (B; n = 5; E, n = 3) were incubated with indicated target MM cells (A, MM1S; B, H929; E, RPMI8226) (E:T = 10:1) in AMG 701 (93.4 pM) or control (ctrl) groups on day 0 (d0). Quantitative FC analysis was used to determine the percentage of differentiated T-cell subtypes: naive (CD45RA+CD62L+), CM (CD45RA−CD62L+), EM (CD45RA−CD62L−), and TE (CD45RA+CD62L−). (A-B, E top panel) Shown are the percentage of CM and EM (means ± SDs) in CD4 and CD8 T subsets (left panel), as well as CD8/CD4 ratios (right panel), at indicated time periods. (C-D) T cells from additional patient samples (C, n = 3; D, n = 5) were coincubated with MM1S (C) or H929 (D) target cells as in panels A and B. (C-D, E bottom panel) FC analysis was used to measure the percentage of Tscm (CD27+CD95+CD45RA+CD62L+) in CD4 and CD8 T subsets. (D) Representative dot plots are shown (left panel) demonstrating gating strategies to define Tscm in CD8 T cells on day 7 in AMG 701 (left) and ctrl (right) groups. (F) T cells from patients (n = 4) were purified after 4-day coculture with H929 target cells (E:T=10:1) in the presence of AMG 701 (93.4 pM). The isolated T cells were then incubated for 24 hours with BCMA-knockdown H929low cells. BCMA ABC was 12 888 vs 273 in the parental H929 vs H929low cells. Shown are the % lysis (means ± SDs) of CD138+ MM target cells (1 and 4, H929low; 2-3, H929) following 24-hour incubation at indicated conditions. Four independent experiments were done at each condition. All data are shown as means ± SDs. *P < .05; **P < .005; ***P < .001; ****P < .0001.
AMG 701 induced T-cell differentiation including scm phenotype, associated with further increases in CD8/CD4 ratios. T cells from donors (A; n = 13) or MM patients (B; n = 5; E, n = 3) were incubated with indicated target MM cells (A, MM1S; B, H929; E, RPMI8226) (E:T = 10:1) in AMG 701 (93.4 pM) or control (ctrl) groups on day 0 (d0). Quantitative FC analysis was used to determine the percentage of differentiated T-cell subtypes: naive (CD45RA+CD62L+), CM (CD45RA−CD62L+), EM (CD45RA−CD62L−), and TE (CD45RA+CD62L−). (A-B, E top panel) Shown are the percentage of CM and EM (means ± SDs) in CD4 and CD8 T subsets (left panel), as well as CD8/CD4 ratios (right panel), at indicated time periods. (C-D) T cells from additional patient samples (C, n = 3; D, n = 5) were coincubated with MM1S (C) or H929 (D) target cells as in panels A and B. (C-D, E bottom panel) FC analysis was used to measure the percentage of Tscm (CD27+CD95+CD45RA+CD62L+) in CD4 and CD8 T subsets. (D) Representative dot plots are shown (left panel) demonstrating gating strategies to define Tscm in CD8 T cells on day 7 in AMG 701 (left) and ctrl (right) groups. (F) T cells from patients (n = 4) were purified after 4-day coculture with H929 target cells (E:T=10:1) in the presence of AMG 701 (93.4 pM). The isolated T cells were then incubated for 24 hours with BCMA-knockdown H929low cells. BCMA ABC was 12 888 vs 273 in the parental H929 vs H929low cells. Shown are the % lysis (means ± SDs) of CD138+ MM target cells (1 and 4, H929low; 2-3, H929) following 24-hour incubation at indicated conditions. Four independent experiments were done at each condition. All data are shown as means ± SDs. *P < .05; **P < .005; ***P < .001; ****P < .0001.
Stem cell–like memory (scm) T cells (Tscm; CD27+CD95+CD45RA+CD62L+) are a subset of long-lived T cells with self-renewal ability and pleiotropic differentiation potential.50-52 This small T-cell subset decreases during progression of MM53 and may influence durability of T-cell–based immunotherapy. We therefore next examined whether AMG 701 induced differentiation of Tscm, in T cells from patients (n = 3, Figure 3C; n = 5, Figure 3D; n = 3, Figure 3E) cocultured with various MM target cell lines (Figure 3C-D and E bottom panel). AMG 701, but not a control BiTE molecule, significantly induced differentiation toward Tscm, as the percentage of Tscm was increased in CD4 and CD8 T subsets starting at day 1 and continuing to day 4 (P < .005) (supplemental Figure 3D-E; Figure 3C-E). Although the percentage of of AMG 701–induced Tscm decreased at day 7, the percentage of of Tscm was still significantly higher in AMG 701–treated vs control T cells.
T cells (healthy donors; n = 3) were purified from 4-day cocultures with AMG 701 and MM target cells and transferred to new cocultures with other MM cell lines. The isolated T cells (n = 3) still induced lysis (83.7% to 98.6%) of these new MM cell lines (n = 2-4; supplemental Figure 3F). Patient T cells (n = 4) isolated from cocultures with H929 target cells after AMG 701 treatment also induced robust lysis of H929low target cells (18.8% to 67.3%; Figure 3F). H929low cells express significantly decreased BCMA levels (47-fold lower, BCMA antibody-binding capacity [ABC] from 12888 to 273), compared with parental H929 cells (supplemental Figure 1B.)
IMiDs synergistically enhanced AMG 701–mediated killing of resistant MM cells
At suboptimal test conditions comprising lower AMG 701 concentrations (<37.4 pM), or lower E:T ratios (<4:1), pretreatment of patient T cells with 1 μM len or pom enhanced AMG 701–mediated TDCC, even against IMiD-resistant H929(R) and RPMI8226 MM cell lines (Figure 4A). Using patient T cells pretreated with len or pom also increased AMG 701–induced H929 MM cell lysis at a shorter incubation time (6 hours), lower E:T ratios (1:1, 2:1, 4:1), and lower AMG 701 doses (<37.4 pM) (supplemental Figure 4A-C). Len- or pom-pretreated patient PBMCs (n = 2) increased AMG 701–mediated lysis of autologous patient MM cells (BCMA ABC of 634 and 1533 for RRMM 1 and RRMM 2, respectively) (P < .05; Figure 4B; supplemental Figure 4D).
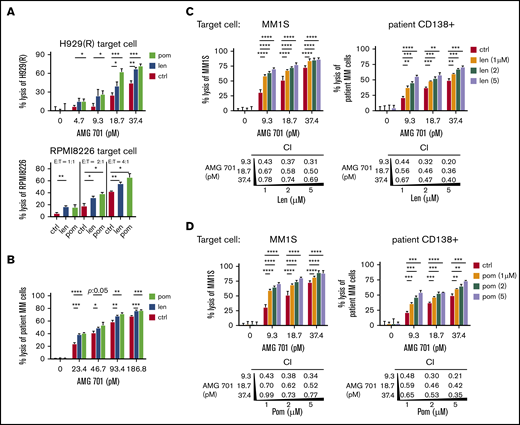
IMiD pretreatment enhanced potency of AMG 701–induced T cytotoxicity against MM cells, including autologous RRMM patient cell lysis, and mediated synergistic cytotoxicity when added concomitantly. (A-B) T cells from 2 patients were pretreated with len (1 μM), pom (1 μM), or control medium (ctrl) for 4 days; drugs were then washed out before 1-day cocultures with MM cells in the presence of AMG 701 at lower E:T ratios and reduced AMG 701 concentrations. FC analysis was used to evaluate the percentage of MM cell lysis. (A) IMiD-resistant H929(R) (top panel) and RPMI8226 (bottom panel) cells were tested at low concentrations of AMG 701 (top panel) or at lower E:T ratios (1:1, 2:1, 4:1) (bottom panel, AMG 701 of 37.4 pM). (B) BMMCs from 2 RRMM patients were cocultured with len- or pom-pretreated PBMCs (PBMC:BMMC = 5:1) from the same patient to evaluate the percent lysis of autologous patient MM cells. len (C) or pom (D) was added at the same time as AMG 701 in 24-hour cocultures of patient T cells (n = 4) with MM1S (left) or autologous patient CD138+ (right) target cells (E:T=5:1). CD138+ cells were coincubated with CD138− cells from the same RRMM patients. Values of CI are shown below each graph. CI < 1 indicates synergism of both drugs. All data are shown as the percent lysis (means ± SDs) from multiple independent experiments (A-B, n = 2; C-D, n = 4) in triplicate at each condition. *P < .05; **P < .01; ***P < .001; ****P < .0001.
IMiD pretreatment enhanced potency of AMG 701–induced T cytotoxicity against MM cells, including autologous RRMM patient cell lysis, and mediated synergistic cytotoxicity when added concomitantly. (A-B) T cells from 2 patients were pretreated with len (1 μM), pom (1 μM), or control medium (ctrl) for 4 days; drugs were then washed out before 1-day cocultures with MM cells in the presence of AMG 701 at lower E:T ratios and reduced AMG 701 concentrations. FC analysis was used to evaluate the percentage of MM cell lysis. (A) IMiD-resistant H929(R) (top panel) and RPMI8226 (bottom panel) cells were tested at low concentrations of AMG 701 (top panel) or at lower E:T ratios (1:1, 2:1, 4:1) (bottom panel, AMG 701 of 37.4 pM). (B) BMMCs from 2 RRMM patients were cocultured with len- or pom-pretreated PBMCs (PBMC:BMMC = 5:1) from the same patient to evaluate the percent lysis of autologous patient MM cells. len (C) or pom (D) was added at the same time as AMG 701 in 24-hour cocultures of patient T cells (n = 4) with MM1S (left) or autologous patient CD138+ (right) target cells (E:T=5:1). CD138+ cells were coincubated with CD138− cells from the same RRMM patients. Values of CI are shown below each graph. CI < 1 indicates synergism of both drugs. All data are shown as the percent lysis (means ± SDs) from multiple independent experiments (A-B, n = 2; C-D, n = 4) in triplicate at each condition. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Various low concentrations of AMG 701 (9.3, 18.7, 37.4 pM) and IMiDs (1, 2, 5 μM) were next added together in 16-hour cocultures of patient T cells (n = 4) with MM1S, H929, or autologous patient CD138+ target cells (Figure 4C-D; supplemental Figure 5). CI47 values <1 at all concentrations indicate synergistic MM cell lysis triggered by AMG 701 combined with len (Figure 4C; supplemental Figure 5B) or with pom (Figure 4D; supplemental Figure 5C).
IMiDs overcame the immunosuppressive effects of BM accessory cells on AMG 701–induced TDCC against MM cells
Effects of IMiDs on AMG 701–induced TDCC of MM cells were further studied in the presence of BM stromal cells (BMSCs) or osteoclasts (OCs) as these BM accessory cells contribute to MM cell growth, survival, drug resistance, and immunosuppression.54-58 IMiD-pretreated T cells significantly increased TDCC against H929 MM cells at low concentrations (<37.4 pM) of AMG 701 (P < .05; Figure 5). Although BMSCs and OCs reduced AMG 701–mediated TDCC of H929 cells, the immunosuppressive effects of these accessory cells were overcome when IMiD-pretreated T cells were used, even at low concentrations of AMG 701. When combined with IMiDs, AMG 701 achieved more MM cell lysis than AMG 701 monotherapy groups, both in the presence or absence of BMSCs or OCs (P < .05; Figure 5).
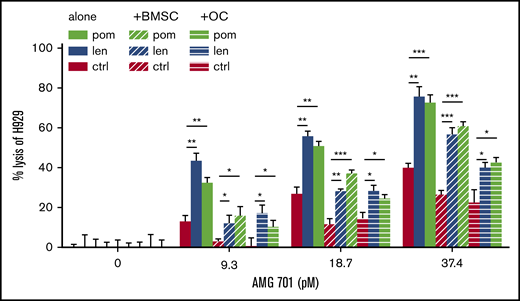
IMiD pretreatment restored AMG 701–induced MM cell lysis in the presence of BMSCs and OCs. T cells were pretreated with len (1 μM), pom (1 μM), or control medium (C) for 4 days. AMG 701–induced TDCC against H929 MM cell lysis was measured in the presence (+) or absence of BMSCs or OCs derived from RRMM patients (n = 3) (E:T = 5:1). The percentage of MM cell lysis (means ± SDs) was evaluated at 1-day coculture. Three independent experiments were done with triplicates for each condition. All results are shown as means ± SDs. *P < .05; **P < .01; ***P < .001.
IMiD pretreatment restored AMG 701–induced MM cell lysis in the presence of BMSCs and OCs. T cells were pretreated with len (1 μM), pom (1 μM), or control medium (C) for 4 days. AMG 701–induced TDCC against H929 MM cell lysis was measured in the presence (+) or absence of BMSCs or OCs derived from RRMM patients (n = 3) (E:T = 5:1). The percentage of MM cell lysis (means ± SDs) was evaluated at 1-day coculture. Three independent experiments were done with triplicates for each condition. All results are shown as means ± SDs. *P < .05; **P < .01; ***P < .001.
AMG 701 and IMiD combination treatment augmented effector cell function via enhanced cell differentiation toward memory cells and reduced immunosuppressive regulators in patient T cells
We next cocultured IMiD-pretreated patient T cells (n > 3) with AMG 701 and MM target cell lines and observed induction of additional increases in the percentage of CM and EM subpopulations in CD4 and CD8 T-cell subsets, as well as increased CD8/CD4 ratios, at day 4 and persisting until day 7 (Figure 6A). In the presence of immunosuppressive OCs, AMG 701 and IMiD combination treatment still induced a higher percentage of CM and EM differentiation in CD4 and CD8 T subsets in a time-dependent manner when compared with AMG 701 alone (Figure 6B). In parallel, IMiD-combination groups showed higher CD8/CD4 ratios than groups treated with AMG 701 alone at day 4 and to an even greater extent at day 7, even in the presence of OCs.
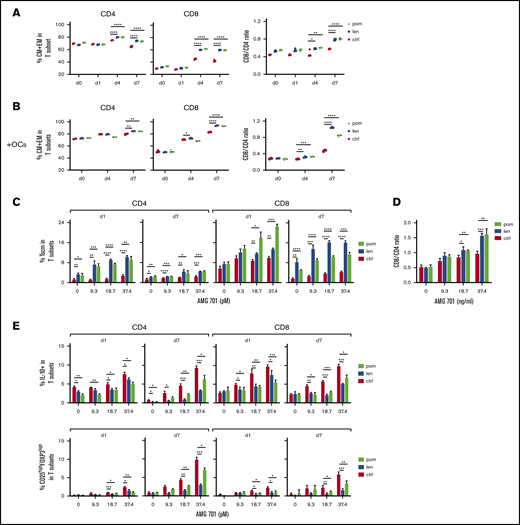
IMiD pretreatment further improved AMG 701–induced T-cell differentiation, CD8/CD4 ratios, and Tscm, and suppressed immune inhibitory regulators in ex vivo cocultures. (A) AMG 701 (46.7 pM) was added into cocultures of H929 with IMiD-pretreated (len or pom, 1 μM) or control (ctrl) patient T cells (n = 3) (E:T = 5:1) on day 0 (d0). Quantitative FC analysis was used to evaluate the percentage of CM and EM in T subsets (left) and the CD8/CD4 ratios (right) at the same time interval. (B) Similar analysis as in panel A was done in the presence of OC using additional T cells (n = 3). (C) Shown are time- and dose-dependency studies on the percentage of Tscm in T subsets (n = 3) in IMiD-pretreated vs ctrl T cells from patients cocultured with H929 MM targets (E:T = 5:1) in the presence of AMG 701. CD8/CD4 ratios at day 7 cocultures were also evaluated (D). (E) Patient T cells (n = 3) treated with 1 μM len, pom, or medium (ctrl) were cocultured with H929 target cells (E:T=5:1) in serial concentrations of AMG 701. Using quantitative FC analysis, the percentage of IL-10+ (top panel) and the percentage of CD25highFOXP3high Treg (bottom panel) in T subsets were evaluated at indicated time intervals. Three independent experiments were done using T cells from 3 individuals. All results are shown as the percent means ± SDs. *P < .05; **P < .01; ***P < .001; ****P < .0001.
IMiD pretreatment further improved AMG 701–induced T-cell differentiation, CD8/CD4 ratios, and Tscm, and suppressed immune inhibitory regulators in ex vivo cocultures. (A) AMG 701 (46.7 pM) was added into cocultures of H929 with IMiD-pretreated (len or pom, 1 μM) or control (ctrl) patient T cells (n = 3) (E:T = 5:1) on day 0 (d0). Quantitative FC analysis was used to evaluate the percentage of CM and EM in T subsets (left) and the CD8/CD4 ratios (right) at the same time interval. (B) Similar analysis as in panel A was done in the presence of OC using additional T cells (n = 3). (C) Shown are time- and dose-dependency studies on the percentage of Tscm in T subsets (n = 3) in IMiD-pretreated vs ctrl T cells from patients cocultured with H929 MM targets (E:T = 5:1) in the presence of AMG 701. CD8/CD4 ratios at day 7 cocultures were also evaluated (D). (E) Patient T cells (n = 3) treated with 1 μM len, pom, or medium (ctrl) were cocultured with H929 target cells (E:T=5:1) in serial concentrations of AMG 701. Using quantitative FC analysis, the percentage of IL-10+ (top panel) and the percentage of CD25highFOXP3high Treg (bottom panel) in T subsets were evaluated at indicated time intervals. Three independent experiments were done using T cells from 3 individuals. All results are shown as the percent means ± SDs. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Transient upregulation of key checkpoint markers was still seen in patient T cells cocultured with MM target cells (MM1S or autologous patient CD138+ cells), in the presence of IMiDs and AMG 701 (supplemental Figure 6). These results suggested that combination treatment did not further induce exhaustion of patient T cells.
IMiDs further enhanced AMG 701–induced Tscm differentiation in patient T cells (n = 3) from days 1 to 7 in cocultures with H929 target cells (P < .05; Figure 6C). Although the percentage of Tscm in CD4 and CD8 T subsets was reduced at day 7 due to AMG 701–induced decreases in the percentage of naive T cells, IMiDs still increased the percentage of Tscm, to a greater extent in CD8 than CD4 T cells (P < .05; Figure 6C). In parallel, IMiDs further enhanced the increase of CD8/CD4 ratios mediated by AMG 701 at day 7 (Figure 6D). T cells (from 4 additional patients) pretreated with IMiDs showed enhanced Tscm differentiation in CD8 vs CD4 T cells and enhanced CD8/CD4 ratios at days 4 and 7, compared with AMG 701 alone (P < .05; supplemental Figure 7).
Interleukin 10 (IL-10) and Tregs mitigate effector T-cell activation, function, and proliferation, thus affecting immunosuppression in MM4,8,57,59,60 ; Tregs also promote MM tumor progression in vivo.59 AMG 701 increased the percentage of IL-10+ T cells in patient samples (n = 3) in a dose- and time-dependent manner. This increase was diminished by cotreatment with IMiDs (P < .05; Figure 6E top panel). IMiDs also decreased AMG 701–induced Treg (CD25highFOXP3high) frequency in CD4 and CD8 T subsets (P < .05; Figure 6E bottom panel). Thus, the combined AMG 701/IMiD treatment decreased AMG 701–induced IL-10+ T-cell and Treg frequencies.
Combined len with AMG 701 treatment continuously inhibited in vivo myeloma cell regrowth
In vivo activity of AMG 701 and len, alone or together, was tested in an advanced-stage H929 xenograft model reconstituted with in vitro–expanded human effector T cells, using doses and treatment schedules that, when given alone, induce an initial response followed by relapse. Female SCID mice (n = 8 per group except n = 5 for vehicle group without human T cells) were xenografted with H929 MM cells. Treatments started on day 15 when tumor volumes (mean ∼260 mm3) showed no significant differences between groups (Figure 7A-B) and continued until the end of the study at day 47. Treatments included administration of len (0.2 mg/kg, intraperitoneally) once daily and/or AMG 701 (0.25 mg/kg, IV) once weekly. Two days after the first drug administration, all 3 regimens significantly inhibited MM tumor growth in mice compared with the vehicle group reconstituted with T cells (supplemental Figure 8; P < .001 at day 24; Figure 7A-B). Most importantly, although tumor regrowth was eventually observed in the monotherapy AMG 701– or len-treated groups, the combination of AMG 701 with len continuously suppressed tumor growth (supplemental Figure 8C; P < .05 from day 26 and P < .001 from day 40 for combination vs either agent alone; Figure 7A-B). Follow-up for 47 days when the experiments were ended has suggested a prolongation in median survival in the combination treatment group vs cohorts treated with either agent alone or control vehicles (vehicle, 18 days; vehicle without T cells, 19 days; AMG 701, 38 days; len, 45 days; AMG 701 plus len, >47 days) (Figure 7C). All 8 mice were alive in the combination treatment group with minimal tumor burden at the end of 47-day follow-up. Thus, combined AMG 701/len treatment significantly inhibited myeloma cell growth in vivo compared with either monotherapy, resulting in longer duration of tumor regression.
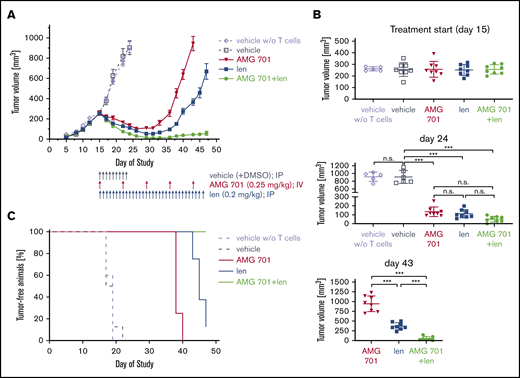
AMG 701 combination treatment with lenalidomide effectively prevented myeloma regrowth in vivo. (A) Head-to-head comparison of AMG 701 and len monotherapy vs combination therapy in the H929 xenograft model in female SCID mice reconstituted with human CD3+ T cells isolated from buffy coats and expanded in vitro. Mice were treated from day 15, when mean tumor volume was 260 mm3 with bolus injections into the peritoneal cavity (vehicle or len) or the lateral tail vein (AMG 701). Control mice reconstituted with or without (w/o) T cells were treated with vehicle once daily from days 15 to 23 (q1dx9). AMG 701 was administered (0.25 mg/kg per administration) once weekly at days 15, 22, 29, 36, and 43; len (0.2 mg/kg per administration) once daily from days 15 to 46 (q1dx32); or both drugs. Eight mice were used in each group, except 5 mice in the vehicle without T-cell group. Values represent mean tumor sizes (mm3) ± SEMs. (B) Symbols indicate individual tumor volumes and lines indicate group mean values ± SDs at days 15, 24, and 43. One-way ANOVA with Tukey multiple comparison test was used for statistics analysis. (C) Using Kaplan-Meier and log-rank analysis, the median overall survival of animals was derived (vehicle, 18 days; vehicle without T cells, 19 days; AMG 701, 38 days; len, 45 days; AMG 701 plus len, >47 days). All 8 mice were alive in the combination therapy group at the end of 47-day follow-up. ***P < .001. n.s., not significant.
AMG 701 combination treatment with lenalidomide effectively prevented myeloma regrowth in vivo. (A) Head-to-head comparison of AMG 701 and len monotherapy vs combination therapy in the H929 xenograft model in female SCID mice reconstituted with human CD3+ T cells isolated from buffy coats and expanded in vitro. Mice were treated from day 15, when mean tumor volume was 260 mm3 with bolus injections into the peritoneal cavity (vehicle or len) or the lateral tail vein (AMG 701). Control mice reconstituted with or without (w/o) T cells were treated with vehicle once daily from days 15 to 23 (q1dx9). AMG 701 was administered (0.25 mg/kg per administration) once weekly at days 15, 22, 29, 36, and 43; len (0.2 mg/kg per administration) once daily from days 15 to 46 (q1dx32); or both drugs. Eight mice were used in each group, except 5 mice in the vehicle without T-cell group. Values represent mean tumor sizes (mm3) ± SEMs. (B) Symbols indicate individual tumor volumes and lines indicate group mean values ± SDs at days 15, 24, and 43. One-way ANOVA with Tukey multiple comparison test was used for statistics analysis. (C) Using Kaplan-Meier and log-rank analysis, the median overall survival of animals was derived (vehicle, 18 days; vehicle without T cells, 19 days; AMG 701, 38 days; len, 45 days; AMG 701 plus len, >47 days). All 8 mice were alive in the combination therapy group at the end of 47-day follow-up. ***P < .001. n.s., not significant.
Discussion
In this study, AMG 701, a novel BCMA-targeting half-life extended BiTE molecule, effectively induced TDCC against MM cell lines and RRMM patient cells. Moreover, in cultures of autologous RRMM BMMC samples containing MM cells and immunosuppressive BM accessory cells, AMG 701 (<46.7 pM) still potently lysed CD138+ patient MM cells, indicating that AMG 701 can induce anti-MM cytotoxicity in the immunosuppressive BM microenvironment in RRMM. Under various test conditions including low AMG 701 concentrations, short coculture time periods, low E:T ratios, and the presence of MM-promoting/immunosuppressive BMSCs or OCs, AMG 701 retained its anti-MM activity. Importantly, AMG 701 effectively depleted MM cells in a mouse MM model when administered on a weekly schedule,46 suggesting a more convenient administration in clinical practice than continuous infusions.23,61
In ex vivo cocultures with MM cells, AMG 701 induced immunomodulatory effects including enhancement of T-cell degranulation and cytolytic cytokine production, promotion of T-cell proliferation, and differentiation from naive to memory phenotypes including CM and EM T cells and Tscm in CD4 and CD8 subsets. Importantly, at the end of 7-day cocultures, CD8/CD4 ratios were consistently increased, even in the presence of immunosuppressive OCs. Moreover, AMG 701 simultaneously induced Tscm in patient T cells, and subsequent T-cell differentiation into effector phenotypes, which may eliminate residual MM cells and prevent relapse. AMG 701 increased the percentage of Tscm in T cells as early as day 1 through day 7. Notably, even though Tscm frequency was reduced at day 7, their frequency at day 7 was still higher in AMG 701–treated vs control-treated groups (to a greater extent in the CD8 vs CD4 subset). In addition, T cells induced to proliferate by AMG 701 in 4-day cocultures demonstrated serial cytotoxicity against other BCMA+ MM cells naive to AMG 701, including those expressing very low cell-surface BCMA. This capacity for serial lysis of target cells may further stimulate an increased repertoire of antigen-experienced antitumor T cells because MM cells express a multitude of tumor antigens on their surface. This “training” or “educating” process mimics the generation of tumor-specific cytotoxic T cells and has been shown to be accelerated by BiTE molecules.54,62,63 Our data show that T cells with memory phenotype or tumor-specific cytotoxic T cells are capable of serial target cell lysis with additional AMG 701 administration, which may result in the development of longer antitumor immunity. These mechanisms may explain the prolonged remission period and high rate of MRD negativity observed in some responders in the first-in-human AMG 420 clinical study.23
Combined AMG 701/IMiD treatment further augmented AMG 701–mediated anti-MM activity by patient T cells in several settings. IMiDs enhanced T-cell function to induce more potent MM cell lysis. IMiDs also promoted formation of immune synapses between immune cells and cancer cells,44,64,65 which may further enhance BiTE-mediated TDCC. Combination AMG 701/IMiD treatment further upregulated AMG 701–induced differentiation of CM and EM T cells, even in the presence of immunosuppressive OCs, leading to further increases in CD8/CD4 ratios. In addition, IMiDs impaired immunosuppressive IL-10+ T cells and Tregs in CD4 and CD8 T-cell subsets induced by AMG 701 monotherapy, starting from day 1 through day 7 in ex vivo cocultures. Thus, AMG 701 in combination with IMiDs further overcame the negative impacts of key inhibitory immune proteins and cells to restore effector cell number and cytolytic function, thereby enhancing and prolonging AMG 701 anti-MM efficacy. IMiDs enhanced AMG 701–induced T-cell activation and memory effector cell and Tscm differentiation at later time points in ex vivo cocultures, suggesting that AMG 701 in combination with IMiDs may prolong duration of response. Importantly, the synergistic anti-MM cytotoxicity of AMG 701 combined with IMiDs shown here may eliminate resistant MRD and thereby extend response durability and prevent disease relapse in patients. The results of our in vivo study demonstrate the superior efficacy of AMG 701 combined with lenalidomide treatment vs monotherapy in achieving better disease control. Such combination strategies may present advantages over monotherapy in clinical practice by delivering deeper and more durable responses with reduced toxicity.
In summary, AMG 701 effectively induced autologous killing of RRMM cells via rapid activation of patient T cells. AMG 701 simultaneously triggered significant proliferation and differentiation of patient T cells, which was associated with significant increases in CD8/CD4 ratios and Tscm. AMG 701 treatment in combination with IMiDs further augmented anti-MM cytotoxicity and immunomodulatoryeffects by increasing memory cells while decreasingimmunosuppressive IL-10 and Treg cells. These data, coupled with superior tumor regression observed after combination therapy in vivo, strongly support AMG 701-based clinical studies, both as monotherapy (NCT03287908) and in combination with IMiDs, toenhance elimination of disease and thereby achieve durable responses in patients with MM.
Data-sharing requests may be e-mailed to the corresponding author, Yu-Tzu Tai, at yu-tzu_tai@dfci.harvard.edu.
Acknowledgments
The authors acknowledge FC assistance from the flow cytometry facility at Dana-Farber Cancer Institute (DFCI). The authors also thank Jiye Liu and Wenjuan Yang at DFCI for helpful technical assistance and discussion for in vitro experiments. The authors thank all other laboratory members as well as the clinical research coordinators at the Jerome Lipper Multiple Myeloma Center and the LeBow Institute for Myeloma Therapeutics of the DFCI for all support and help in providing primary tumor specimens for this study.
This work was supported in part by grants from the National Institutes of Health, National Cancer Institute Specialized Programs of Research Excellence (SPORE) P50 CA100707, P01CA155258, and RO1 CA 207237. This work was supported in part by the Miriam and Sheldon G. Adelson Medical Research Foundation. K.C.A. is an American Cancer Society Clinical Research Professor. This study was funded by Amgen.
Authorship
Contribution: K.C.A. and Y.-T.T. conceived and designed the study; S.-F.C., L.L., L.X., and Y.-T.T. developed the methodology; L.L., S.-F.C., L.X., T.Y., Y.L., K.W., and P.A.H. acquired data (provided reagents, facilities, etc); T.A. provided reagents and materials; S.-F.C., L.L., L.X., T.Y., Y.L., K.W., P.A.H., and Y.-T.T. analyzed and interpreted data (statistical analysis, biostatistics analysis); J.W., K.M., M.F., and T.A. designed animal work and collected and analyzed data (biostatistics analysis); N.M. and K.C.A. provided and managed patient samples; S.-F.C., K.C.A., and Y.-T.T. wrote, reviewed, and/or revised the manuscript; and K.C.A. and Y.-T.T. supervised the study.
Conflict-of-interest disclosure: J.W., K.M., M.F., and T.A. are employees of Amgen and have stock and/or stock interests in Amgen. N.M. serves on advisory boards to Millennium-Takeda, Celgene, and Novartis. K.C.A. serves on advisory boards to Celgene, Millennium-Takeda, Bristol-Myers Squibb, Gilead Sciences, Janssen, and Sanofi-Aventis and is a scientific founder of OncoPep and C4 Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Yu-Tzu Tai, Department of Medical Oncology, Dana-Farber Cancer Institute, M551, 450 Brookline Ave, Boston, MA 02215; e-mail: yu-tzu_tai@dfci.harvard.edu; and Kenneth C. Anderson, Department of Medical Oncology, Dana-Farber Cancer Institute, M557, 450 Brookline Ave, Boston, MA 02215; e-mail: kenneth_anderson@dfci.harvard.edu.

