

Key Points
The dual PI3Kδ/CK1ε inhibitor umbralisib exhibits unique immunomodulatory effects on CLL T cells.
Umbralisib disables WNT signaling through inhibition of CK1ε and exerts less detrimental effects on Treg immunosuppressive function.

Abstract
The in-clinic phosphatidylinositol 3-kinase (PI3K) inhibitors idelalisib (CAL-101) and duvelisib (IPI-145) have demonstrated high rates of response and progression-free survival in clinical trials of B-cell malignancies, such as chronic lymphocytic leukemia (CLL). However, a high incidence of adverse events has led to frequent discontinuations, limiting the clinical development of these inhibitors. By contrast, the dual PI3Kδ/casein kinase-1-ε (CK1ε) inhibitor umbralisib (TGR-1202) also shows high rates of response in clinical trials but has an improved safety profile with fewer severe adverse events. Toxicities typical of this class of PI3K inhibitors are largely thought to be immune mediated, but they are poorly characterized. Here, we report the effects of idelalisib, duvelisib, and umbralisib on regulatory T cells (Tregs) on normal human T cells, T cells from CLL patients, and T cells in an Eμ-TCL1 adoptive transfer mouse CLL model. Ex vivo studies revealed differential effects of these PI3K inhibitors; only umbralisib treatment sustained normal and CLL-associated FoxP3+ human Tregs. Further, although all 3 inhibitors exhibit antitumor efficacy in the Eμ-TCL1 CLL model, idelalisib- or duvelisib-treated mice displayed increased immune-mediated toxicities, impaired function, and reduced numbers of Tregs, whereas Treg number and function were preserved in umbralisib-treated CLL-bearing mice. Finally, our studies demonstrate that inhibition of CK1ε can improve CLL Treg number and function. Interestingly, CK1ε inhibition mitigated impairment of CLL Tregs by PI3K inhibitors in combination treatment. These results suggest that the improved safety profile of umbralisib is due to its role as a dual PI3Kδ/CK1ε inhibitor that preserves Treg number and function.
Introduction
In mammalian cells, phosphatidylinositol 3-kinase (PI3K) signaling occurs upon binding of a regulatory subunit (p85 or p55) with a catalytic subunit (p110α, β, δ, or γ). This leads to activation of AKT and downstream effectors that control cell growth, survival, and metabolism.1 In cells of hematopoietic origin, expression of the p110 PI3Kδ catalytic subunit predominates and plays essential roles in B-cell development, survival, and function.2,3 For example, in mature B cells, PI3K signaling is initiated downstream of B-cell receptor signaling, where it controls activation and differentiation responses,2,3 and mice lacking a catalytically active p110δ are viable but have impaired B-cell responses.4 Notably, p110δ-deficient mice also display impaired T-cell functions and develop an inflammatory bowel syndrome that has been attributed to defective regulatory T cells (Tregs) and that is manifest as marked lymphocyte infiltrates into intestinal tissues.4 Given these observations, the role of PI3K signaling in T-cell differentiation, function, and immune tolerance has also been studied.5 The p110γ catalytic subunit is also known to play important roles in PI3K signaling downstream of G-protein–coupled receptors in T cells and myeloid cells.6,7 PI3K inhibitors targeting the p110γ isoform have been investigated in the context of cancers and autoimmunity. However, it is unclear how p110δ and p110γ subunits interact to enable PI3K signaling in T cells, what specific functions they each control, and whether they can act in a compensatory manner in cells lacking or inhibited in 1 isoform.
Dependence on PI3K signaling is conserved in malignant B cells, including chronic lymphocytic leukemia (CLL) B cells,1,8 and this has led to the recent clinical development and US Food and Drug Administration (FDA) approval of PI3K inhibitors for the treatment of B-cell malignancies.9 However, there is a lack of robust data characterizing the effects of these inhibitors in secondary target cell populations. Because of the importance of PI3K signaling in multiple immune cell types, PI3K inhibitors targeting ≥1 catalytic isoform can have unexpected immunomodulatory effects, which may vary depending on each inhibitor’s selectivity, potency, and off-target profile.
The immunomodulatory properties of PI3K inhibitors are significant in the context of hematological malignancies and other cancers. For example, although the antitumor effects of the PI3K inhibitors idelalisib and duvelisib are clear, the high incidence of immune-mediated severe adverse events (AEs) in clinical trials among patients with B-cell malignancies, including colitis, pneumonitis, and hepatotoxicity, has caused high rates of discontinuation that have limited the development of a potentially long-term beneficial treatment.10 In contrast, treatment with umbralisib, a next-generation dual inhibitor of PI3Kδ and casein kinase-1-ε (CK1ε),11 has shown an improved safety profile that is characterized by less severe AEs, even with long-term follow-up.12 The 50% inhibitory concentrations of idelalisib, duvelisib, and umbralisib against p110 isoforms in a cell-free enzymatic assay have been published previously.13
It has been postulated that T cells may modulate PI3K inhibitor–related AEs. Increased inflammatory cytokine levels and decreased Treg numbers have been associated with the development of AEs in CLL patients on idelalisib,14 and younger treatment-naive patients, who likely have more robust immune systems, were at higher risk for developing toxicities. In another study, CLL patients on umbralisib in combination with ublituximab and pembrolizumab retained numbers and percentage of circulating Tregs over time, and immune-mediated severe AEs were not increased above what was expected for umbralisib or pembrolizumab alone.15 There is a clear need to understand the effects of pharmacological PI3K inhibition in T cells and other immune populations.
In the context of hematological malignancies, T cells may develop a phenotype supporting malignant progression, differing substantially from that of healthy T cells. For example, CLL patients often harbor low numbers of T cells that display markers of exhaustion, are less proliferative, and have inverted T helper 1 (Th1)/Th2 cell and CD4/CD8 ratios; they can also have increased Treg numbers.16 Here, we characterized the immunomodulatory effects of clinically available PI3Kδ inhibitors and report marked and differential effects of these drugs on Tregs in the setting of murine CLL. Importantly, analyses revealed similar anti-CLL efficacy, but favorable toxicity profiles, for the dual PI3K/CK1ε inhibitor umbralisib vs idelalisib and duvelisib, as well as that umbralisib was associated with improved function and sustained numbers of Tregs, supporting the clinical utility of this agent in the treatment of CLL.
Methods
Cell culture
Cells were cultured in RPMI 1640 plus 10% fetal bovine serum, 5% penicillin-streptomycin, 5% nonessential amino acids, and 1% MycoZap and incubated at 37°C with 5% CO2. Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood using Ficoll-Paque density gradient separation. Cells were isolated using magnetic separation kits (STEMCELL Technologies, Cambridge, MA), and viability assays were conducted (CellTiter-Blue Cell Viability Assay; Promega, Madison, WI).
CLL patient samples
PBMCs were obtained from CLL patients. All participants gave written Institutional Review Board–approved informed consent for blood to be used for research (#CR6_Pro00000316). Blood was collected at the H. Lee Moffitt Cancer Center and Research Institute. CLL patients were diagnosed according to the 2018 guidelines of the International Workshop on Chronic Lymphocytic Leukemia.17 Patient characteristics are detailed in Table 1, and patients were treatment naive at the time of sample collection.
Table of CLL patient characteristics
| Age, y | Sex | Cytogenetics (FISH) | CD38+ | ZAP70 | IGHV status | |
|---|---|---|---|---|---|---|
| CLL 1 | 75 | F | Trisomy 12 | Positive | Negative | Mutated |
| CLL 2 | 72 | F | 13q del | Positive | Negative | Mutated |
| CLL 3 | 46 | F | 13q del | Unknown | Negative | Mutated |
| CLL 4 | 41 | F | Trisomy 12 | Positive | Negative | Unknown |
| CLL 5 | 53 | M | Normal | Positive | Positive | Unmutated |
| CLL 6 | 72 | M | 13q del | Negative | Negative | Mutated |
| CLL 7 | 63 | M | 13q del | Negative | Positive | Unmutated |
| CLL 8 | 54 | M | 13q del | Negative | Unknown | Mutated |
| CLL 9 | 73 | M | 13q del | Negative | Negative | Mutated |
| CLL 10 | 74 | M | 13q del | Positive | Negative | Mutated |
| CLL 11 | 51 | M | 11q del | Negative | Positive | Unmutated |
| CLL 12 | 71 | M | Trisomy 12 | Positive | Negative | Mutated |
| CLL 13 | 69 | M | Normal | Positive | Negative | Mutated |
| CLL 14 | 72 | M | Normal | Positive | Negative | Unmutated |
| CLL 15 | 67 | F | 13q del, 17p del | Negative | Negative | Mutated |
| CLL 16 | 74 | M | 13q del | Negative | Negative | Mutated |
| CLL 17 | 50 | F | Normal | Negative | Negative | Mutated |
| Age, y | Sex | Cytogenetics (FISH) | CD38+ | ZAP70 | IGHV status | |
|---|---|---|---|---|---|---|
| CLL 1 | 75 | F | Trisomy 12 | Positive | Negative | Mutated |
| CLL 2 | 72 | F | 13q del | Positive | Negative | Mutated |
| CLL 3 | 46 | F | 13q del | Unknown | Negative | Mutated |
| CLL 4 | 41 | F | Trisomy 12 | Positive | Negative | Unknown |
| CLL 5 | 53 | M | Normal | Positive | Positive | Unmutated |
| CLL 6 | 72 | M | 13q del | Negative | Negative | Mutated |
| CLL 7 | 63 | M | 13q del | Negative | Positive | Unmutated |
| CLL 8 | 54 | M | 13q del | Negative | Unknown | Mutated |
| CLL 9 | 73 | M | 13q del | Negative | Negative | Mutated |
| CLL 10 | 74 | M | 13q del | Positive | Negative | Mutated |
| CLL 11 | 51 | M | 11q del | Negative | Positive | Unmutated |
| CLL 12 | 71 | M | Trisomy 12 | Positive | Negative | Mutated |
| CLL 13 | 69 | M | Normal | Positive | Negative | Mutated |
| CLL 14 | 72 | M | Normal | Positive | Negative | Unmutated |
| CLL 15 | 67 | F | 13q del, 17p del | Negative | Negative | Mutated |
| CLL 16 | 74 | M | 13q del | Negative | Negative | Mutated |
| CLL 17 | 50 | F | Normal | Negative | Negative | Mutated |
F, female; FISH, fluorescence in situ hybridization; M, male.
Treg polarization and suppression assay
Naive CD4+ T cells were isolated from human whole blood using a Human CD4+ T cell isolation kit and cultured in Treg-polarizing conditions (50 IU/mL human interleukin-2 [IL-2], 5 ng/mL human transforming growth factor-β1, ImmunoCult CD3/CD28 stimulation; STEMCELL Technologies) for 5 days. Tregs were washed, treated with inhibitors for 24 hours, and cultured with autologous responder CD4+ T cells labeled with CellTrace Violet (ThermoFisher Scientific, Waltham, MA) at a 1:5 (Treg/CD4+) ratio for 5 days. CellTrace Violet dilution was measured via flow cytometry.
PI3K and CK1ε inhibitors
Idelalisib (CAL-101),18 duvelisib (IPI-145),19 and PF-4800567 (a CK1ε-selective small-molecule inhibitor)20 were obtained commercially (Selleckchem.com, Houston, TX). Umbralisib (TGR-1202)12 was kindly provided by TG Therapeutics. Inhibitors were dissolved and stored in dimethyl sulfoxide (DMSO) at −20°C.
CLL mouse model
A total of 25 × 106 leukemic splenocytes from female Eμ-TCL121 mice was adoptively transferred via the tail vein into female wild-type (6-8 weeks old) syngeneic C57BL/6 mice to induce CLL disease. Leukemic Eμ-TCL1 mice were defined as >6 months of age with >70% B cells in total lymphocytes in peripheral blood by flow analysis. Disease induction was confirmed by significantly elevated white blood cell count within 3 weeks of adoptive transfer. Inhibitors were administered as a suspension in Tween-20 and 90% methylcellulose/deionized water once daily by oral gavage at a final concentration of 100 mg/kg.At the end point, animals were euthanized for collection of organs. Peripheral blood and spleen were used for immunophenotyping analysis, other in vitro assays, and to assess tumor burden. Liver and gastrointestinal tract from tumor-bearing recipients were collected for histology scoring analysis of toxicity.
GVHD mouse model
A total of 5 × 106 bone marrow cells isolated from femurs of female wild-type C57Bl/6 mice was adoptively transferred into female wild-type BALB/c mice that were irradiated (900 rad). Treatment was administered via oral gavage beginning 1 day after adoptive transfer. The incidence of graft-versus-host disease (GVHD) was tracked by weight loss over time.
Histological analysis of immune-mediated toxicity
Adoptive transfer Eμ-TCL1 mice were euthanized after 21 days of oral treatment with umbralisib, idelalisib, or duvelisib (100 mg/kg). Intestinal tissue (small intestine and colon) and liver were collected from each mouse and fixed in formaldehyde. Tissues were embedded in paraffin and stained with hematoxylin and eosin. Liver tissue was observed in a blinded analysis for inflammatory foci and hepatocyte injury as parameters of immune-mediated toxicity. Small intestine and colon tissue were examined in a blinded analysis for immune infiltrate, shortened villi, and denuded mucosa. Depending on the frequency and severity of these parameters, each mouse was assigned a toxicity score for each tissue type as follows: 0-1, absent/mild; 2, moderate; and 3, severe. Scores for each tissue type were summed for an overall toxicity grade per mouse, and overall toxicity grades were averaged for comparison of each treatment group.
Flow cytometry
Immunophenotyping analysis was performed via flow cytometry. All markers were gated according to fluorescence minus 1 or isotype controls. Acquisition was performed on a BD LSR II cytometer (BD Biosciences, Franklin Lakes, NJ), and data were analyzed with FlowJo software v10.1 (TreeStar, Ashland, OR).
Surface and intracellular flow cytometry
Cells were stained for surface antigens with the indicated antibodies, according to the manufacturer’s dilution, for 1 hour at room temperature in FACS buffer (phosphate-buffered saline with 2% fetal bovine serum). Viability staining was accomplished using LIVE/DEAD fixable dyes or 4′,6-diamidino-2-phenylindole exclusion (ThermoFisher Scientific). To detect intracellular proteins, cells were fixed following surface staining using a Transcription Factor Buffer Set (BD Biosciences, San Diego, CA), as per the manufacturer’s protocol. The following anti-human or anti-mouse antibodies were used: CD3, CD19, CD4, CD8, CD25, CD127, FoxP3, PD-1, CTLA-4 (BD Biosciences, Franklin Lakes, NJ), CD38, and β-catenin (both from BioLegend, San Diego, CA). Absolute cell counts were determined using AccuCheck Counting Beads (Life Technologies, Frederick, MD), according to the manufacturer’s protocol.
Phospho-flow cytometry analyses
Isolated human or mouse T cells were stimulated with functional grade plate-bound human or mouse anti-CD3/CD28 (CD3 at 10 μg/mL and CD28 at 2 μg/mL; eBioscience, San Diego, CA) for 20 minutes. Cells were immediately fixed in 1% formaldehyde for 15 minutes at 37°C and permeabilized in 90% ice-cold methanol for 1 hour. Cells were washed and stained in FACS buffer with anti-AKT (pS473) (BD Biosciences, Franklin Lakes, NJ) antibody, according to the manufacturer’s instructions. Median fluorescence intensity (MFI) values were normalized to isotype control, and percentage change was calculated using DMSO or vehicle as 100%. Acquisition was performed on an iQue Screener PLUS (IntelliCyt, Albuquerque, NM) and analyzed with accompanying software.
Cytokines
Cytokine Bead Array kits (BD Biosciences, Franklin Lakes, NJ) were used, according to the manufacturer’s protocol, to detect secreted human or mouse Th1/Th2 cytokines from supernatant of CD3+ T cells 24 hours after stimulation with anti-CD3/anti-CD28. Acquisition was performed on a BD FACSCanto cytometer and analyzed with FCAP array software version 3.0 (both from BD Biosciences, Franklin Lakes, NJ).
Statistics
Statistical significance between data sets was determined by an unpaired, 2-tailed, or Student t test if data were normally distributed or by a Mann-Whitney U unpaired test if data were not normally distributed. For groups of 3 or more, ordinary 1-way analysis of variance (ANOVA), followed by Tukey’s multiple-comparisons test, was used if data were normally distributed; a Kruskal-Wallis test was used if data were not normally distributed. For correlation studies, linear regression was performed on data sets. A P value < .05 was considered significant. Analyses were conducted using GraphPad Prism software v7.
Animal studies approval
All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee at the University of South Florida/Comparative Medicine.
Results
Suppressive T-cell phenotypes are differentially modulated by PI3K inhibitors
To initially assess potential immunomodulatory effects of PI3K inhibitors, T cells were isolated from healthy donor or CLL PBMCs. Isolated T cells were cultured with PI3K inhibitors idelalisib, duvelisib, or umbralisib or vehicle (DMSO) for 24 hours at various doses, and viability was measured by a CellTiter-Blue Cell Viability Assay. Treatment with these inhibitors did not compromise survival of normal or CLL T cells at doses of 0.1 to 10 μM (Figure 1A); hence, this dose range was selected for all in vitro experiments. To confirm whether these agents were on target, phosphorylated AKT (pAKT) was used as a readout of PI3K pathway inhibition in normal and CLL T cells. As predicted, reductions in levels of pAKT were observed and were similar among all inhibitors (Figure 1B).
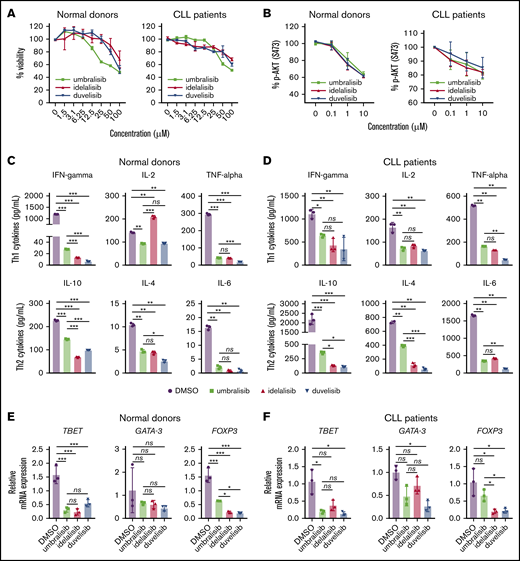
PI3K inhibitors impair normal and CLL human T-cell survival and function. (A) CellTiter-Blue assay of T-cell viability after 24 hours of treatment with the indicated concentrations of PI3K inhibitors. (B) Inhibition of PI3K signaling in normal or CLL patient CD3+ T cells isolated from human PBMCs was measured by assessing levels of pS473-AKT after 24 hours of incubation with inhibitors, followed by stimulation with anti-CD3/anti-CD28. Graphs display mean + standard deviation (SD) compiled from all samples; n = 3 normal donors and n = 4 CLL patient samples. (C-D) Cytokine bead array analysis of Th1/Th2 cytokines secreted from human CD3+ normal or CLL T cells incubated with the indicated inhibitors (1 μM, for 24 hours), following anti-CD3/anti-CD28 stimulation. (E-F) The expression of Th transcription factors was assessed by qRT-PCR in CD3+ T cells incubated with the indicated inhibitors (1 μM, 24 hours), followed by anti-CD3/anti-CD28 stimulation (24 hours). Graph displays ΔΔCT for each transcription factor. Data in (C-F) are representative of 3 independent experiments, using a unique donor for each experiment. Graphs display mean + SD of technical triplicates from a representative donor. *P < .05, **P < .005, ***P < .0005, ordinary 1-way ANOVA. ns, not statistically significant.
PI3K inhibitors impair normal and CLL human T-cell survival and function. (A) CellTiter-Blue assay of T-cell viability after 24 hours of treatment with the indicated concentrations of PI3K inhibitors. (B) Inhibition of PI3K signaling in normal or CLL patient CD3+ T cells isolated from human PBMCs was measured by assessing levels of pS473-AKT after 24 hours of incubation with inhibitors, followed by stimulation with anti-CD3/anti-CD28. Graphs display mean + standard deviation (SD) compiled from all samples; n = 3 normal donors and n = 4 CLL patient samples. (C-D) Cytokine bead array analysis of Th1/Th2 cytokines secreted from human CD3+ normal or CLL T cells incubated with the indicated inhibitors (1 μM, for 24 hours), following anti-CD3/anti-CD28 stimulation. (E-F) The expression of Th transcription factors was assessed by qRT-PCR in CD3+ T cells incubated with the indicated inhibitors (1 μM, 24 hours), followed by anti-CD3/anti-CD28 stimulation (24 hours). Graph displays ΔΔCT for each transcription factor. Data in (C-F) are representative of 3 independent experiments, using a unique donor for each experiment. Graphs display mean + SD of technical triplicates from a representative donor. *P < .05, **P < .005, ***P < .0005, ordinary 1-way ANOVA. ns, not statistically significant.
To assess potential effects on T-cell functions, cytokine secretion was measured from the supernatant of T cells incubated with the 3 PI3K inhibitors (Figure 1C-D). Overall, Th1 and Th2 cytokines were decreased following treatment with PI3K inhibitors, yet IL-10 production was higher following umbralisib treatment compared with idelalisib or duvelisib treatment of normal and CLL T cells.
The potential effects of these PI3K inhibitors on T-cell fate was assessed by examining Th lineage transcription factors using quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR). Expression of Tbet (Th1) was markedly suppressed by all 3 inhibitors, whereas expression of the Th2 determination factor GATA-3 was unaffected in normal samples and suppressed by umbralisib and duvelisib in CLL samples (Figure 1E-F). Idelalisib or duvelisib treatment also led to profound suppression of FOXP3 (Treg) expression, whereas FOXP3 levels were more sustained in umbralisib-treated T cells. Thus, PI3K inhibitors impair Th cell differentiation, and idelalisib or duvelisib treatment has more detrimental effects on FoxP3+ Tregs than does treatment with umbralisib.
Given these findings, the effects of these PI3K inhibitors on anti-CD3/anti-CD28–induced differentiation of total T cells from normal or CLL donors were assessed by flow cytometry. Although PI3K treatment did not affect total T-cell count or the CD4/CD8 ratio (Figure 2A), total Treg numbers were reduced in a dose-dependent fashion following treatment with idelalisib or duvelisib (gating strategy is shown in supplemental Figure 1). In contrast, Treg numbers were more sustained in umbralisib-treated T cells (Figure 2A; supplemental Figure 2A). Interestingly, the cell surface expression of the immunosuppressive checkpoint molecules PD-1 and CTLA-4 on Tregs was sustained in umbralisib-treated samples but was suppressed by idelalisib or duvelisib treatment (Figure 2B; supplemental Figure 2B). These findings were confirmed in a cohort of 6 normal donor samples and 6 CLL patient samples; the percentage of Tregs, as well as surface levels of PD-1 and CTLA-4 on Tregs, was relatively preserved in umbralisib-treated normal or CLL patient T cells (Figure 2C-D; supplemental Figure 3).
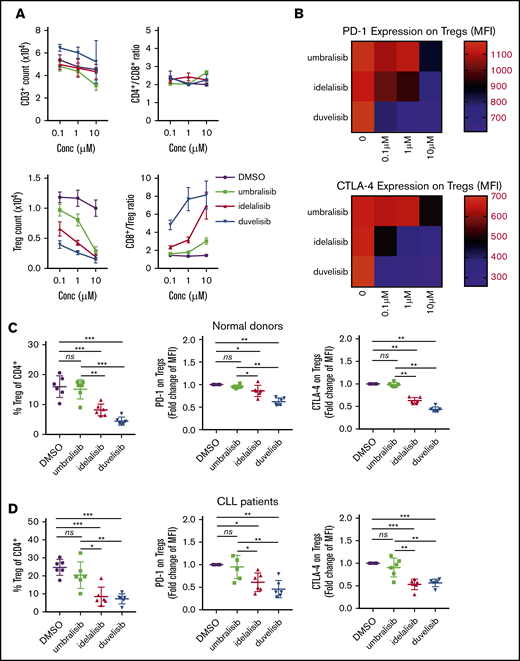
PI3K inhibitors differentially impair normal and CLL Tregs in a similar manner. (A) CD3+ T cells were isolated from normal donor PBMCs and cultured with the indicated concentrations of PI3K inhibitors in dose titration in the presence of anti-CD3/anti-CD28 for 72 hours. Graphs display mean of technical triplicates + standard deviation (SD) from 1 representative donor. (B) PD-1 and CTLA-4 expression on Tregs in (A). Heat map depicts average MFI of technical triplicates from 1 representative donor. (C) CD3+ T cells were isolated from normal donor PBMCs (n = 6) and cultured with PI3K inhibitors (1 μM, 72 hours) in the presence of anti-CD3/anti-CD28. (D) CD3+ T cells were isolated from previously frozen CLL patient PBMCs (n = 6) and cultured with PI3K inhibitors (1 μM, 72 hours) in the presence of anti-CD3/anti-CD28. Immunophenotyping was performed via flow cytometry. PD-1 and CTLA-4 MFIs were normalized to DMSO to stabilize variance. Graphs display mean + SD. *P < .05, **P < .005, ***P < .0005, ordinary 1-way ANOVA.
PI3K inhibitors differentially impair normal and CLL Tregs in a similar manner. (A) CD3+ T cells were isolated from normal donor PBMCs and cultured with the indicated concentrations of PI3K inhibitors in dose titration in the presence of anti-CD3/anti-CD28 for 72 hours. Graphs display mean of technical triplicates + standard deviation (SD) from 1 representative donor. (B) PD-1 and CTLA-4 expression on Tregs in (A). Heat map depicts average MFI of technical triplicates from 1 representative donor. (C) CD3+ T cells were isolated from normal donor PBMCs (n = 6) and cultured with PI3K inhibitors (1 μM, 72 hours) in the presence of anti-CD3/anti-CD28. (D) CD3+ T cells were isolated from previously frozen CLL patient PBMCs (n = 6) and cultured with PI3K inhibitors (1 μM, 72 hours) in the presence of anti-CD3/anti-CD28. Immunophenotyping was performed via flow cytometry. PD-1 and CTLA-4 MFIs were normalized to DMSO to stabilize variance. Graphs display mean + SD. *P < .05, **P < .005, ***P < .0005, ordinary 1-way ANOVA.
To assess the potential effects of PI3K inhibitors on Treg function, we performed a Treg suppression assay, in which normal donor Tregs were treated with inhibitors for 24 hours, washed, and cocultured with autologous responder CD4+ T cells. Treatment with duvelisib and idelalisib impaired the suppressive capacity of Tregs, whereas umbralisib-treated Tregs retained more of their suppressive capacity (Figure 3).
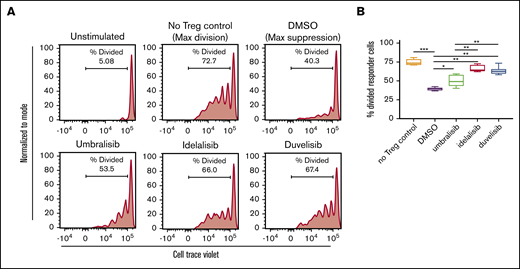
Treg suppressive capacity is only modestly impaired by umbralisib treatment. (A) Naive CD4+ T cells were isolated from normal donor PBMCs and cultured under Treg-polarizing conditions for 5 days. Tregs were washed and pretreated with PI3K inhibitors (1 μM, 24 hours) and then washed and cocultured with autologous CellTrace Violet–labeled responder T cells. Cell division was assessed by dilution of CellTrace Violet in responder cells 5 days after beginning coculture. Line graphs depict division of responder T cells from 1 representative donor. (B) Box and whisker plots show quantification of suppression assay (n = 6) as mean + standard deviation. *P < .05, **P < .005, ***P < .0005, ordinary 1-way ANOVA.
Treg suppressive capacity is only modestly impaired by umbralisib treatment. (A) Naive CD4+ T cells were isolated from normal donor PBMCs and cultured under Treg-polarizing conditions for 5 days. Tregs were washed and pretreated with PI3K inhibitors (1 μM, 24 hours) and then washed and cocultured with autologous CellTrace Violet–labeled responder T cells. Cell division was assessed by dilution of CellTrace Violet in responder cells 5 days after beginning coculture. Line graphs depict division of responder T cells from 1 representative donor. (B) Box and whisker plots show quantification of suppression assay (n = 6) as mean + standard deviation. *P < .05, **P < .005, ***P < .0005, ordinary 1-way ANOVA.
Differential effects of PI3K inhibitor treatment on CLL Tregs
To assess whether differential regulation of Tregs by PI3K inhibitors was also manifest in the setting of CLL, we used the Eμ-TCL1 adoptive transfer mouse model of CLL. As predicted, oral administration of idelalisib, duvelisib, or umbralisib led to marked reductions in pAKT in Eμ-TCL1 T cells (Figure 4A). Pharmacokinetic analyses established that all 3 inhibitors were detected in plasma at 1 hour postdosing (Figure 4B) and remained detectable for up to 16 hours. Following oral administration of idelalisib, duvelisib, or umbralisib (21 days, once daily, 100 mg/kg), CLL burden (CD19+ CD5+ B cells) in peripheral blood was significantly decreased compared with vehicle-treated mice (Figure 4C). Reduction in tumor burden was confirmed in splenocytes (data not shown). Total CD3+ T-cell numbers were also lower in inhibitor-treated mice vs vehicle-treated mice (Figure 4D), yet the CD4/CD8 ratio was decreased only in duvelisib-treated mice (Figure 4E). Notably, total Treg numbers were higher in the umbralisib group than in the idelalisib or duvelisib group (Figure 4F). The expression of several surface markers involved in Treg suppressive function (transforming growth factor-β1, GITR, CD73)22 was also higher in the umbralisib-treated cohort compared with idelalisib- and duvelisib-treated mice, suggesting that umbralisib-treated Tregs retain functional capacity in the CLL setting (Figure 4G). Finally, to assess functional capacity, the 4 cohorts of Eμ-TCL1 mice were euthanized, and splenic CD3+ T cells were activated ex vivo. Notably, IL-10 production was more sustained in T cells isolated from umbralisib-treated Eμ-TCL1 mice (Figure 4H).
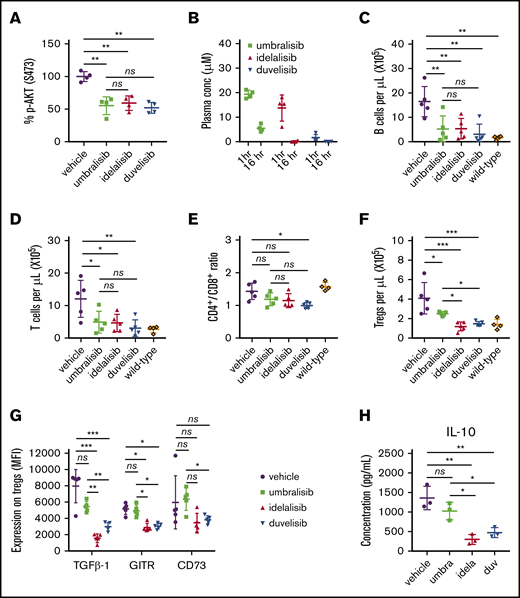
Treatment with PI3K inhibitors differentially impairs Tregs in Eμ-TCL1 mice. (A) In vivo activity of PI3K inhibitors was assessed by monitoring levels of pS473-AKT in splenic T cells after 3 days of treatment (100 mg/kg, once daily, oral gavage) in wild-type mice (n = 4 per group). (B) Eμ-TCL1 CLL-bearing recipient mice were randomized and treated for 21 days with the indicated inhibitors (100 mg/kg, once daily, n = 5 mice per group). The plasma concentration of each PI3K inhibitor was measured by high-performance liquid chromatography after collection of peripheral blood at 1 hour and 16 hours after oral gavage. (C) Antitumor efficacy of inhibitors was determined by quantifying CLL B cells in peripheral blood following 21 days of treatment (100 mg/kg, n = 5 mice per group). (D) Total CD3+ T-cell count in peripheral blood in the animals treated in (C). CD4/CD8 ratio (E) and Treg count (F) in peripheral blood in the animals treated in (C). (G-H) Total CD3+ splenic T cells from the mice treated in (C) were stimulated ex vivo for 24 hours with anti-CD3/CD28. Expression of functional markers on the surface of Tregs was determined by flow cytometry, and IL-10 secretion was measured in the supernatant by cytometric bead array. MFIs of functional markers were normalized to fluorescence minus 1 controls. Graphs show mean + standard deviation. Data are representative of 3 independent in vivo experiments. *P < .05, **P < .005, ***P < .0005, ordinary 1-way ANOVA.
Treatment with PI3K inhibitors differentially impairs Tregs in Eμ-TCL1 mice. (A) In vivo activity of PI3K inhibitors was assessed by monitoring levels of pS473-AKT in splenic T cells after 3 days of treatment (100 mg/kg, once daily, oral gavage) in wild-type mice (n = 4 per group). (B) Eμ-TCL1 CLL-bearing recipient mice were randomized and treated for 21 days with the indicated inhibitors (100 mg/kg, once daily, n = 5 mice per group). The plasma concentration of each PI3K inhibitor was measured by high-performance liquid chromatography after collection of peripheral blood at 1 hour and 16 hours after oral gavage. (C) Antitumor efficacy of inhibitors was determined by quantifying CLL B cells in peripheral blood following 21 days of treatment (100 mg/kg, n = 5 mice per group). (D) Total CD3+ T-cell count in peripheral blood in the animals treated in (C). CD4/CD8 ratio (E) and Treg count (F) in peripheral blood in the animals treated in (C). (G-H) Total CD3+ splenic T cells from the mice treated in (C) were stimulated ex vivo for 24 hours with anti-CD3/CD28. Expression of functional markers on the surface of Tregs was determined by flow cytometry, and IL-10 secretion was measured in the supernatant by cytometric bead array. MFIs of functional markers were normalized to fluorescence minus 1 controls. Graphs show mean + standard deviation. Data are representative of 3 independent in vivo experiments. *P < .05, **P < .005, ***P < .0005, ordinary 1-way ANOVA.
Treg number is associated with incidence of immune-mediated toxicities in CLL mice
B-cell cancer patients treated with idelalisib, duvelisib, and umbralisib can suffer from immune-mediated toxicities.13 To investigate this phenomenon, we collected tissues susceptible to AEs from Eμ-TCL1 mice treated with the PI3K inhibitors. In a blinded histological analysis, each mouse was assigned a toxicity grade based on histological features of the small intestine, colon, and liver (criteria are summarized in Table 2). Idelalisib- and duvelisib-treated Eμ-TCL1 mice accrued the highest toxicity grades, whereas umbralisib-treated Eμ-TCL1 mice displayed a much lower incidence of AEs (Figure 5A-B). This result was reminiscent of available toxicity data derived from clinical use of these inhibitors.12,14
Criteria for histological analysis of immune-mediated toxicity
| Organ site | Criteria for immune-mediated toxicity |
|---|---|
| Liver | Inflammatory foci, hepatocyte injury |
| Small intestine/colon | Immune infiltrate, shortened villi, denuded mucosa |
| Organ site | Criteria for immune-mediated toxicity |
|---|---|
| Liver | Inflammatory foci, hepatocyte injury |
| Small intestine/colon | Immune infiltrate, shortened villi, denuded mucosa |
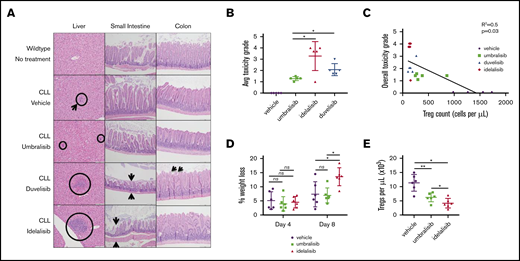
Analysis of immune-mediated toxicity in CLL murine model. (A) Representative histology findings in liver, small intestine, and colon of Eμ-TCL1 CLL-bearing recipient mice treated as described in Figure 4. Circles and arrows depict sites showing signs of toxicity (original magnification, ×20; hematoxylin & eosin stain). (B) Average toxicity grade per group (n = 5 mice per group, from Figure 4). (C) Linear regression of peripheral Treg count vs incidence of toxicity reveals an inverse correlation. (D) Percentage weight loss in the GVHD murine model normalized to weight prior to sublethal irradiation (inhibitors were administered at 100 mg/kg, once daily, oral gavage; n = 6 mice per group). (E) Submandibular bleeds were collected from each mouse on day 8, and immunophenotyping was performed by flow cytometry to detect peripheral CD4+ CD25HI FoxP3+ Tregs. Data are mean + standard deviation. Data are representative of 3 independent in vivo experiments. *P < .05, **P < .005, ***P < .0005, ordinary 1-way ANOVA.
Analysis of immune-mediated toxicity in CLL murine model. (A) Representative histology findings in liver, small intestine, and colon of Eμ-TCL1 CLL-bearing recipient mice treated as described in Figure 4. Circles and arrows depict sites showing signs of toxicity (original magnification, ×20; hematoxylin & eosin stain). (B) Average toxicity grade per group (n = 5 mice per group, from Figure 4). (C) Linear regression of peripheral Treg count vs incidence of toxicity reveals an inverse correlation. (D) Percentage weight loss in the GVHD murine model normalized to weight prior to sublethal irradiation (inhibitors were administered at 100 mg/kg, once daily, oral gavage; n = 6 mice per group). (E) Submandibular bleeds were collected from each mouse on day 8, and immunophenotyping was performed by flow cytometry to detect peripheral CD4+ CD25HI FoxP3+ Tregs. Data are mean + standard deviation. Data are representative of 3 independent in vivo experiments. *P < .05, **P < .005, ***P < .0005, ordinary 1-way ANOVA.
It has been postulated that deleterious effects on Tregs may be involved in the regulation of immune-mediated toxicities that are characteristic of PI3K inhibitors.13 Given the differences in peripheral Treg number and function following treatment of Eμ-TCL1 mice with these 3 PI3K inhibitors, a correlation analysis was performed to determine whether Treg number was related to incidence of toxicities. Interestingly, a reduced number of peripheral Tregs positively correlated with overall toxicity grade in Eμ-TCL1 treated mice (Figure 5C).
To assess the effects of these PI3K inhibitors on Treg suppressive capacity in vivo, a major histocompatibility complex–mismatched GVHD mouse model (C57BL/6 bone marrow donor → BALB/c recipient) was used in which functional Tregs are known to dampen the course of GVHD.23 Following engraftment, recipient mice were treated orally with umbralisib, idelalisib, or vehicle and were followed for the course of their disease. Notably, GVHD was exacerbated in mice treated with idelalisib, whereas mice treated with umbralisib did not show any difference from the vehicle cohort (Figure 5D). Finally, umbralisib-treated mice had twofold greater numbers of Tregs than did idelalisib-treated mice (Figure 5E). Thus, umbralisib treatment does not compromise Treg suppressive functions in vivo.
CK1ε inhibition protects Tregs from the detrimental effects of PI3K inhibition
Umbralisib is the only PI3K inhibitor known to inhibit CK1ε.11 Anti-CK1ε activity has been implicated in this inhibitor’s antitumor activity toward B-cell lymphomas as a result of downregulation of c-MYC.11 CK1ε drives canonical Wnt signaling in mammalian cells,24 where it suppresses the activity of the β-catenin destruction complex and promotes β-catenin nuclear translocation and the transcription of WNT/β-catenin target genes.25 Accordingly, CK1ε silencing suppresses the expression and activity of β-catenin and its target genes.26 WNT signaling has been implicated in the differentiation and function of T-cell subsets,27 yet the roles of CK1ε in T-cell function have not been investigated.
To assess whether CK1ε inhibition by umbralisib contributes to sparing CLL Treg number and function, we used a CK1ε-selective small molecule inhibitor, PF-4800567, which does not inhibit the closely related isoform CK1δ or the more distantly related family member CK1α at the concentrations used.20 Treatment with PF-4800567 did not compromise CLL T-cell survival (supplemental Figure 4). PF-4800567 and umbralisib led to dose-dependent reductions in protein expression of β-catenin, which is easily detectable by flow cytometry, in CLL patient T cells (Figure 6A). Levels of WNT effector gene Axin2 transcripts in normal T cells were also suppressed by PF-4800567 or umbralisib treatment but not by idelalisib treatment (Figure 6B). These results suggested that inhibition of CK1ε by umbralisib was necessary for suppression of β-catenin and Axin2 expression.
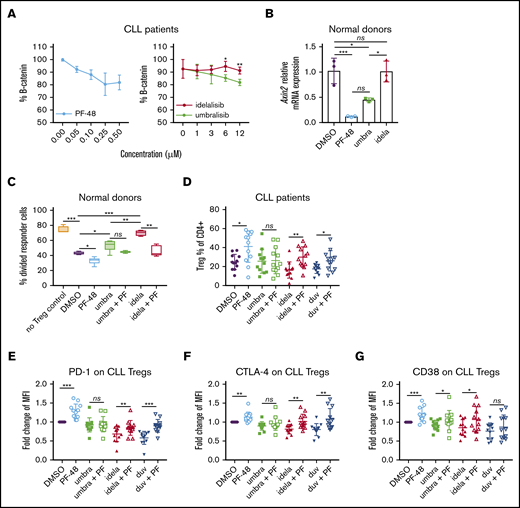
Effects of CK1ε and dual PI3K/CK1ε inhibition on normal and CLL human Tregs. (A) Protein expression of β-catenin was detected by intracellular flow cytometry in CLL patient T cells that were isolated by magnetic separation from previously frozen PBMCs and treated with PF-4800567, a CK1ε selective inhibitor, or PI3K inhibitors for 24 hours (n = 3). Graphs display mean + standard deviation (SD). (B) Normal human T cells were treated for 24 hours with the indicated inhibitors and then lysed in TRIzol Reagent. Axin2 messenger RNA levels were measured by qRT-PCR. Graph displays mean ΔΔCT. Error bars denote technical triplicates of a representative donor typical of 3 independent experiments. (C) A Treg suppression assay was performed as in Figure 3, following treatment with PI3K inhibitors (1 μM) with or without PF-4800567 (250 nM). (D-G) CLL patient T cells were isolated and cultured in Treg-polarizing conditions for 5 days in the presence of PF-4800567 (250 nM) and PI3K inhibitors (1 μM). Flow cytometry was performed to assess Treg percentage (D) and immunosuppressive markers (E-G) (n = 12). Graphs display mean + SD. *P < .05, **P < .005, ***P < .0005, ordinary 1-way ANOVA.
Effects of CK1ε and dual PI3K/CK1ε inhibition on normal and CLL human Tregs. (A) Protein expression of β-catenin was detected by intracellular flow cytometry in CLL patient T cells that were isolated by magnetic separation from previously frozen PBMCs and treated with PF-4800567, a CK1ε selective inhibitor, or PI3K inhibitors for 24 hours (n = 3). Graphs display mean + standard deviation (SD). (B) Normal human T cells were treated for 24 hours with the indicated inhibitors and then lysed in TRIzol Reagent. Axin2 messenger RNA levels were measured by qRT-PCR. Graph displays mean ΔΔCT. Error bars denote technical triplicates of a representative donor typical of 3 independent experiments. (C) A Treg suppression assay was performed as in Figure 3, following treatment with PI3K inhibitors (1 μM) with or without PF-4800567 (250 nM). (D-G) CLL patient T cells were isolated and cultured in Treg-polarizing conditions for 5 days in the presence of PF-4800567 (250 nM) and PI3K inhibitors (1 μM). Flow cytometry was performed to assess Treg percentage (D) and immunosuppressive markers (E-G) (n = 12). Graphs display mean + SD. *P < .05, **P < .005, ***P < .0005, ordinary 1-way ANOVA.
To test whether CK1ε inhibition affects Treg biology, a Treg suppression assay was conducted using normal human Tregs pretreated with vehicle, PF-4800567, PI3K inhibitors, or combinations of PI3K inhibitors plus PF-4800567. As expected, the repressive effects of umbralisib on Tregs were more modest than were those of idelalisib (Figure 6C; supplemental Figure 4). Further, treatment with PF-4800567 improved the suppressive activity of Tregs. Notably, dual treatment with PF-4800567 impaired the deleterious effects of idelalisib on the suppressive functions of Tregs (Figure 6C; supplemental Figure 4). Other investigators have previously reported that activation of WNT signaling can disrupt Treg differentiation and function.28 Therefore, we asked whether regulation of Tregs by CK1ε inhibition was dependent on WNT signaling. Concomitantly, anti-CD3/anti-CD28–induced Treg differentiation was impaired by stimulation of WNT signaling with lithium chloride, a known WNT pathway agonist,28,29 and this effect was partially abrogated by treatment with the CK1ε inhibitor PF-4800567 (supplemental Figure 5).
To test whether these findings were also manifest in the context of CLL, CLL patient T cells were isolated and cultured in the presence of inhibitors under Treg-polarizing conditions and then examined by flow cytometry. Induction of Tregs was increased by treatment with PF-4800567 compared with DMSO control (Figure 6D). Further, cotreatment with PF-4800567 partially impaired the reductions in Treg induction expected of idelalisib and duvelisib (Figure 6D). Finally, expression of immunosuppressive markers PD-1, CTLA-4, and CD38, which are typically highly expressed on functional Tregs, was reduced by idelalisib and duvelisib treatment. However, dual treatment with PF-4800567 partially impaired these reductions (Figure 6E-G). These findings are consistent with the notion that CK1ε inhibition by umbralisib impairs reductions in Treg number and function that are provoked by the PI3K inhibitors idelalisib and duvelisib.
Discussion
PI3K signaling is dysregulated in many cancers, including solid tumors and lymphoid malignancies. Activating mutations occur in multiple genes that direct PI3K signaling, including PI3KCA, PTEN, AKT, and mTOR.30 Mutations also occur in downstream effectors of PI3K signaling, including antiapoptosis BCL family proteins.31,32 Furthermore, mutations and the resulting pathway activation are associated with acquired resistance to therapeutic agents.33,34 Based on these data, PI3K inhibitors were developed for clinical application. Several antitumor mechanisms have been described, including inhibition of prosurvival signaling, disruption of microenvironment signals, and enhanced antitumor immunity.13 Accordingly, there has been a drive to develop isoform-selective inhibitors to enhance selectivity in targeted cell types and reduce toxicity. Here, we assessed the immunomodulatory effects of PI3K inhibitors idelalisib, duvelisib, and umbralisib, which differ in their clinical safety profiles.13
Idelalisib was approved by the FDA in 2014 for the treatment of patients with relapsed follicular B-cell non-Hodgkin lymphoma, for small lymphocytic leukemia patients who had received ≥2 prior systemic therapies. It was also approved for relapsed CLL patients in combination with rituximab.35 Although high response rates and increased overall survival were demonstrated in patients treated with idelalisib, severe AEs were well documented,10 and frontline trials for CLL patients were halted because of the high incidence of AEs. Notably, disruption of the immune system has been linked to these AEs.36 For example, idelalisib treatment was reported to impair production of IL-6, IL-10, and tumor necrosis factor-α by activated CD3+ T cells and interferon-γ in activated NK cells.18 However, in vitro studies of idelalisib did not characterize effects on CLL T cells; considering the differences in T-cell subsets and function in CLL,16 we reasoned that the immunomodulatory effects of PI3K inhibitors could be more profound in this context. Although our data agree with prior studies showing no cytotoxicity of idelalisib against human T cells and a general reduction in Th1/Th2 cytokine secretion (Figure 1), the data clearly reveal profound reductions in Treg number and function in normal and CLL human T cells following in vitro treatment (Figure 2).
Clinical data indicate a correlation between inhibition of p110δ and autoimmune toxicities.14,36 A decreased percentage of Tregs has been associated with the development of toxicities in patients treated with idelalisib.14 To thoroughly address the immunomodulatory effects of the clinically available PI3Kδ inhibitors, we directly compared the effects of several PI3K inhibitors in a validated mouse model of CLL. Interestingly, peripheral Treg number correlates negatively with incidence of autoimmune toxicity (Figure 5), suggesting that functional Tregs are indispensable to protect against autoimmune toxicity. Thus, peripheral Treg count and/or IL-10 secretion from peripheral blood T cells could be explored as potential predictive biomarkers for autoimmune toxicities in patients who are treated with idelalisib or other PI3K inhibitors.
Duvelisib is an inhibitor of p110δ and p110γ19 ; however, it is more potent against p110δ than is idelalisib in cell-free enzymatic assays.12 Duvelisib has been tested as a monotherapy and in combination with other agents to treat B-cell malignancies in numerous trials, and it is approved by the FDA for CLL and follicular lymphoma after failing 2 lines of therapy.37 Its toxicity profile was similar to idelalisib and included transaminitis, neutropenia, colitis, and pneumonitis.13 Further, in vitro studies have demonstrated cytotoxicity of duvelisib toward human CLL T cells and reductions in IL-2, tumor necrosis factor-α, and interferon-γ production by normal healthy human T cells.38 Concordant with these findings, duvelisib treatment of normal and CLL T cells provokes reductions in Th1/Th2 cytokines (Figure 1), as well as in Treg number and function (Figures 2-3). Further, despite duvelisib plasma levels being lower than those of idelalisib and umbralisib in the Eμ-TCL1 CLL model (Figure 4), the reductions in pAKT in T cells were similar, the decreases in Treg numbers were more profound, and there was a higher incidence of toxicity compared with vehicle- or umbralisib-treated mice (Figure 5).
Umbralisib has enhanced selectivity for inhibiting p110δ over other PI3K isoforms and is unique in that it also inhibits CK1ε.11 Umbralisib has been tested in clinical trials encompassing several B-cell malignancies, with high response rates and progression-free survival similar to those previously demonstrated with idelalisib. Notably, however, umbralisib has an improved safety profile, with fewer incidences of immune-mediated AEs and discontinuations due to toxicities.12 CLL patients treated with umbralisib were reported to retain circulating Tregs over time.15 Concordant with these findings, in the Eμ-TCL1 model, umbralisib-treated mice experienced the lowest level of immune-mediated toxicity among the 3 treatments (Figure 5), and this was associated with higher numbers of functional Tregs. Although sufficient long-term data from patients treated with umbralisib that assess correlations between lymphocyte subsets and incidence of severe AEs are not yet available, a prediction from the findings presented herein is that the improved safety profile of umbralisib will correlate with sustained Tregs in treated patients. The differential impact of umbralisib on Tregs compared with other PI3Kδ inhibitors has several clinical implications. First, the cancer cell–intrinsic inhibition of PI3Kδ and CK1ε by umbralisib uniquely provides multiple strategies of antitumor efficacy in B-cell malignancies. Second, it is interesting to note that lack of Treg impairment by umbralisib may negate potential Treg-mediated antitumor effects in B-cell malignancies, as well as in the setting of immunotherapy for solid tumors. However, the effect of umbralisib on other immune populations that may modulate tumor survival or antitumor efficacy, such as the myeloid subset, remains to be explored.
Janovska and colleagues recently explored inhibition of CK1δ/CK1ε as a therapeutic target in CLL.39 Their study reported that treatment with the dual CK1δ/CK1ε inhibitor PF-670462 elicited an antitumor benefit in the Eμ-TCL1 model. Because umbralisib is unique in its ability to selectively inhibit CK1ε, we reasoned that this might account for the improved toxicity profile of this PI3K inhibitor. To our surprise, treatment with the CK1ε-selective inhibitor PF-4800567 increased the suppressive capacity of normal Tregs, as well as partially impaired the detrimental effect of idelalisib treatment on Treg suppression. Treatment with PF-4800567 also increased the percentage of Tregs in CLL patients and improved expression of suppressive markers when combined with PI3K inhibitors ex vivo (Figure 6). The effects of PF-4800567 and umbralisib were associated with suppression of β-catenin and Axin2 in T cells, indicating that WNT signaling is downregulated by these inhibitors but not by idelalisib. Interestingly, other investigators have recently shown that WNT pathway stimulation in Tregs provokes TCF-1–dependent induction of IL2 transcription and the suppression of FoxP3 transcriptional activity, resulting in decreased Treg function.28 Collectively, these findings support a model whereby impairment of Treg fate and function by PI3Kδ inhibition can be counteracted with CK1ε inhibition. These findings offer an explanation for the immunomodulatory differences seen in clinical applications of the dual PI3Kδ/CK1ε inhibitor umbralisib compared with other PI3K inhibitors, and they suggest that this circuit may be operational for a broad spectrum of malignancies treated with PI3K inhibitors. They also suggest that once a CK1ε inhibitor safety candidate is developed, a CLL clinical trial testing its utility as a single agent or as a combination therapeutic with PI3K inhibitors should be considered.
Data sharing requests should be sent to Eva Sahakian (eva.sahakian@moffitt.org).
Acknowledgments
The authors thank the staff and expert service of the Flow Cytometry Core, Tissue Core, Analytical Microscopy Core, and Comparative Medicine Core of the H. Lee Moffitt Cancer Center and Research Institute. They also thank Carlo Croce (Ohio State University) for kindly providing the Eμ-TCL1 mouse model. Finally, they thank Sonya J. Smyk (H. Lee Moffitt Cancer Center and Research Institute), for editorial support; she was not compensated beyond her regular salary.
This work was supported in part through a Sponsored Research Agreement with TG Therapeutics (J.P.-I. and E.S.), by the Cortner-Couch Endowed Chair for Cancer Research of the University of South Florida (J.L.C.), by funds from National Institutes of Health National Cancer Center Core Support Grant P30-CA076292, and by funds from the State of Florida to the H. Lee Moffitt Cancer Center and Research Institute.
Authorship
Contribution: K.M. designed and conducted research, analyzed and interpreted data, and wrote the manuscript; J.J.P., A.A., M.M.-V., W.G., K.L.B., and R.F. conducted research; K.J. analyzed data; J.L.C., W.R.R., D.R.D., A.M., D.M., and H.P.M. served as advisors; J.L.C. edited the manuscript; and E.S. and J.P.-I. conceived and designed research, interpreted data, and edited the manuscript.
Conflict-of-interest disclosure: J.P.-I. and E.S. have received research funding from TG Therapeutics. J.P.-I has received honoraria from Gilead for acting as a consultant and for serving on its Advisory Board. H.P.M. and D.M. are employees of and have equity ownership in TG Therapeutics. The remaining authors declare no compering financial interests.
Correspondence: Eva Sahakian, Department of Malignant Hematology, H. Lee Moffitt Cancer Center and Research Institute, 12902 Magnolia Dr, Tampa, FL 33612; e-mail: eva.sahakian@moffitt.org.

