

Key Points
This RIC regimen paired with single-unit cord blood results in >90% event-free survival effective for 20 nonmalignant conditions.
The regimen obviates the need for pharmacokinetic dose adjustments, rendering it applicable even in resource-limited centers and countries.

Abstract
Children with many inherited nonmalignant disorders can be cured or their condition alleviated by hematopoietic stem cell transplantation (HSCT). Umbilical cord blood (UCB) units are a rapidly available stem cell source and offer great flexibility in HLA matching, allowing nearly uniform access to HSCT. Although reduced-intensity conditioning (RIC) regimens promise decreased treatment-related morbidity and mortality, graft failure and infections have limited their use in chemotherapy-naive patients. We prospectively evaluated a novel RIC regimen of alemtuzumab, hydroxyurea, fludarabine, melphalan, and thiotepa with a single-unit UCB graft in 44 consecutive patients with inborn errors of metabolism, immunity, or hematopoiesis. In addition, 5% of the UCB graft was re-cryopreserved and reserved for cord donor leukocyte infusion (cDLI) posttransplant. All patients engrafted at a median of 15 days posttransplant, and chimerism was >90% donor in the majority of patients at 1-year posttransplant with only 1 secondary graft failure. The incidence of grade II to IV graft-versus-host disease (GVHD) was 27% (95% confidence interval [CI], 17-43) with no extensive chronic GVHD. Overall survival was 95% (95% CI, 83-99) and 85% (95% CI, 64-93) at 1 and 5 years posttransplant, respectively. No significant end-organ toxicities were observed. The use of cDLI did not affect GVHD and showed signals of efficacy for infection control or donor chimerism. This RIC transplant regimen using single-unit UCB graft resulted in outstanding survival and remarkably low rates of graft failure. Implementation of the protocol not requiring pharmacokinetic monitoring would be feasible and applicable worldwide for children with inherited disorders of metabolism, immunity, or hematopoiesis. This trial was registered at www.clinicaltrials.gov as #NCT01962415.
Introduction
Hematopoietic stem cell transplantation (HSCT) from a healthy donor can cure or ameliorate pathology in a broad spectrum of nonmalignant disorders (NMDs), including primary immunodeficiency diseases, hemoglobinopathies, bone marrow failure syndromes, and inborn errors of metabolism (IEM),1-4 by replacing defective red blood cells or leukocytes or by releasing previously missing enzymes.4-6 Although myeloablative conditioning (MAC) was initially used to show efficacy, the benefits of HSCT have been reported following reduced-intensity conditioning (RIC) even in settings of partial host stem cell recovery, called mixed donor–recipient chimerism.7,8 RIC regimens have also shown decreased morbidity and treatment-related mortality (TRM) compared with MAC regimens7-9 ; however, their widespread use has been limited by a higher incidence of graft failure in chemotherapy-naive patients undergoing unrelated umbilical cord blood (UCB) transplantation (UCBT) compared with those who have received previous chemotherapy.7,9-11 UCBT is uniquely suited for children with NMD given its rapid availability and freedom from strict HLA matching requirements, thus making UCBT theoretically possible for >95% of pediatric patients regardless of their ethnic background.12,13 Historically, a high proportion of patients who may benefit from allogeneic HSCT are not referred for transplantation, as TRM for UCBT has remained in the range of 10% to 30% at 1 year for MAC and RIC regimens, and many patients require second transplantation.14 Procedure-related morbidity may also limit referral from geneticists, hematologists, and immunologists, particularly for patients who exhibit advanced disease–specific symptoms.
We sought to design an UCBT trial that is safe and suitable for every child with NMDs other than chromosomal breakage syndromes or severe combined immunodeficiency, who may benefit from less-intensive conditioning regimens. We built on our pilot experience using hydroxyurea, alemtuzumab, fludarabine, melphalan, and thiotepa. This pilot trial (#NCT00744692) was the first to formally show noninferior engraftment and survival after RIC UCBT compared with MAC regimens (at >90% and 77%, respectively) along with very low end-organ toxicity. Nevertheless, due to mortality related to adenovirus and cytomegalovirus (CMV) infections, this trial failed to show superiority compared with MAC regimens.15 The current trial was designed to preserve the benefits of reduced organ toxicity seen in the previous trial while improving survival through enhanced immune recovery in all disease categories. We postulated that immune recovery had been hampered by lympholytic levels of alemtuzumab peri-transplantation; therefore, the cumulative dose was reduced for all disease categories and administered closer to transplantation to shorten the pretransplant course. In addition, a personalized alemtuzumab de-escalation strata was implemented driven by the primary diagnosis as it may modify the risk of rejection, along with pre-UCBT lymphocyte numbers that alemtuzumab is aimed to delete. Further de-escalation was instituted for recent infection by potentially life-threatening viruses in which rapid donor lymphocyte recovery is desirable even if graft-versus-host disease (GVHD) risk was increased. We also tested the hypothesis that cord donor leukocyte infusion (cDLI) from a small, recryopreserved fraction of the thawed UCB graft could jump-start T-cell reconstitution in high-risk individuals if performed a few weeks after UCBT, when the alemtuzumab level has declined, to accelerate resolution of viral infections and/or bolster donor T-cell chimerism.
Patients and methods
Patients
The first 44 consecutive patients with 20 genetically distinct NMDs (Table 1) underwent UCBT at UPMC Children’s Hospital of Pittsburgh between September 2011 and December 2018 on an institutional review board–approved, prospective clinical trial (www.clinicaltrials.gov: #NCT01962415). Patients with severe combined immunodeficiency syndrome or chromosomal breakage syndromes were excluded, as they would benefit from additional reductions in chemotherapy intensity. The first 15 patients received an identical chemotherapy regimen before ClinicalTrials.gov registration. Because there was no difference in the conditioning regimen or patient characteristics (supplemental Table 1), all 44 patients were analyzed together. Informed consent was obtained from legal guardians of each patient. All patients had at least 1 year of follow-up, and all were chemotherapy naive.
Patient and graft characteristics (N = 44)
| Variables | Value |
|---|---|
| Patient characteristics | |
| Age, median (range), y | 1.7 (0.4-16.6) |
| Weight, median (range), kg | 12 (6-74) |
| Male sex, n (%) | 27 (61) |
| Race, n (%) | |
| White | 29 (66) |
| African American | 8 (18) |
| Hispanic | 3 (7) |
| Asian | 1 (2) |
| Alaska Native | 1 (2) |
| 2 or more races | 2 (5) |
| Diagnosis, n (%) | |
| Krabbe disease | 13 (30) |
| Metachromatic leukodystrophy | 7 (16) |
| Sickle cell disease | 3 (7) |
| Gaucher disease | 2 (5) |
| Hunter syndrome (MPS type II) | 2 (5) |
| MHC class II deficiency | 2 (5) |
| Osteopetrosis | 2 (5) |
| XLP2 | 2 (5) |
| β-thalassemia | 1 (2) |
| Cartilage-hair hypoplasia | 1 (2) |
| Chédiak-Higashi syndrome | 1 (2) |
| Combined immunodeficiency with multiple intestinal atresias | 1 (2) |
| Diamond-Blackfan anemia | 1 (2) |
| GM3 synthase deficiency | 1 (2) |
| Hemophagocytic lymphohistiocytosis | 1 (2) |
| Hurler syndrome (MPS type IH) | 1 (2) |
| Severe congenital neutropenia | 1 (2) |
| Tay-Sachs disease | 1 (2) |
| X-linked adrenoleukodystrophy | 1 (2) |
| CMV serostatus, seropositive | 16 (36) |
| Graft characteristics | |
| HLA match (of 6), n (%) | |
| 6 | 16 (36) |
| 5 | 19 (43) |
| 4 | 9 (21) |
| HLA match (of 8), n (%) | |
| 8 | 8 (18) |
| 7 | 10 (23) |
| 6 | 14 (32) |
| 5 | 10 (23) |
| 4 | 2 (5) |
| TNC/kg, median (range), ×107 | 9.1 (2.3-24.8) |
| CD34/kg, median (range), ×105 | 3.31 (0.92-9.24) |
| Sex mismatch, n (%) | 22 (50) |
| ABO mismatch, n (%) | 28 (64) |
| Variables | Value |
|---|---|
| Patient characteristics | |
| Age, median (range), y | 1.7 (0.4-16.6) |
| Weight, median (range), kg | 12 (6-74) |
| Male sex, n (%) | 27 (61) |
| Race, n (%) | |
| White | 29 (66) |
| African American | 8 (18) |
| Hispanic | 3 (7) |
| Asian | 1 (2) |
| Alaska Native | 1 (2) |
| 2 or more races | 2 (5) |
| Diagnosis, n (%) | |
| Krabbe disease | 13 (30) |
| Metachromatic leukodystrophy | 7 (16) |
| Sickle cell disease | 3 (7) |
| Gaucher disease | 2 (5) |
| Hunter syndrome (MPS type II) | 2 (5) |
| MHC class II deficiency | 2 (5) |
| Osteopetrosis | 2 (5) |
| XLP2 | 2 (5) |
| β-thalassemia | 1 (2) |
| Cartilage-hair hypoplasia | 1 (2) |
| Chédiak-Higashi syndrome | 1 (2) |
| Combined immunodeficiency with multiple intestinal atresias | 1 (2) |
| Diamond-Blackfan anemia | 1 (2) |
| GM3 synthase deficiency | 1 (2) |
| Hemophagocytic lymphohistiocytosis | 1 (2) |
| Hurler syndrome (MPS type IH) | 1 (2) |
| Severe congenital neutropenia | 1 (2) |
| Tay-Sachs disease | 1 (2) |
| X-linked adrenoleukodystrophy | 1 (2) |
| CMV serostatus, seropositive | 16 (36) |
| Graft characteristics | |
| HLA match (of 6), n (%) | |
| 6 | 16 (36) |
| 5 | 19 (43) |
| 4 | 9 (21) |
| HLA match (of 8), n (%) | |
| 8 | 8 (18) |
| 7 | 10 (23) |
| 6 | 14 (32) |
| 5 | 10 (23) |
| 4 | 2 (5) |
| TNC/kg, median (range), ×107 | 9.1 (2.3-24.8) |
| CD34/kg, median (range), ×105 | 3.31 (0.92-9.24) |
| Sex mismatch, n (%) | 22 (50) |
| ABO mismatch, n (%) | 28 (64) |
MHC, major histocompatibility complex; MPS, mucopolysaccharidosis; TNC, total nucleated cell; XLP2, X-linked lymphoproliferative syndrome type 2.
Donors
UCB grafts were selected based on HLA-A and HLA-B intermediate-resolution and HLA-DRB1 allele-level typing. Although allele-level typing was also examined, unit selection was primarily based on intermediate-resolution HLA typing with a 5 of 6 match preferred over a 4 of 6 match and targeting a precryopreserved total nucleated cell and CD34+ cell dose ≥3 × 107/kg and ≥1.5 × 105/kg, respectively. For IEM patients, candidate units meeting these parameters were evaluated for enzyme activity, where feasible, and units with low enzyme activity were excluded. Donor-specific antibodies were tested in 31 patients.
Conditioning regimen
Risk-stratified alemtuzumab dosing was 0, 0.5, or 1 mg/kg IV on day −13 following a test dose for tolerability of 0.2 mg/kg on day −14. Patients with underlying conditions of lymphopenia and/or preexisting viral infections at risk to recur were stratified to lower alemtuzumab dosing (supplemental Table 2). Oral hydroxyurea 30 mg/kg per day started between day −20 and −15 through day −5; fludarabine 30 mg/m2 per day (or 1 mg/kg per day, whichever dose was lower) IV on days −9 to −5; melphalan 70 mg/m2 per day IV on days −4 to −3; and thiotepa 200 mg/m2 on day −2. Patients with transfusion-dependent anemias started hydroxyurea on day −22 and received alemtuzumab with a test dose on day −21 and dose per stratum on days −20 and −19 (supplemental Figure 1).
GVHD prophylaxis
All patients received GVHD prophylaxis with tacrolimus (continuous infusion target level, 12-16 ng/mL) and mycophenolate mofetil (MMF) 15 mg/kg IV q8h from day −3. Tacrolimus was converted to twice-daily oral dosing prior to discharge with target trough levels of 8 to 12 ng/mL. In patients with no active GVHD, MMF was discontinued after day 28, and tacrolimus taper was initiated at day 100 in patients without GVHD. Those with active viral infections were permitted to undergo faster taper of MMF. Acute GVHD was graded according to 1994 consensus criteria.16
Supportive care
Patients were hospitalized before alemtuzumab infusion. Fungal prophylaxis with IV caspofungin was replaced with oral voriconazole near discharge. Bacterial prophylaxis with levofloxacin was given starting on day 0. After December 2013, all patients with CMV seropositivity and/or polymerase chain reaction positivity received ganciclovir prophylaxis from day −12 to −1, followed by acyclovir or foscarnet daily. Patients at risk for herpes simplex virus or varicella-zoster virus received acyclovir from day 1. Beginning after administration of alemtuzumab, viral monitoring was performed twice weekly for adenovirus, 1 to 2 times weekly for CMV, and every 2 weeks for Epstein-Barr virus with pharmacologic intervention upon confirmed viremia. Pneumocystis jirovecii prophylaxis was with trimethoprim-sulfamethoxazole pretransplantation and IV pentamidine after day 28. Patients received IV immunoglobulin 500 mg/kg every 2 weeks post-alemtuzumab until month 2 and for an immunoglobulin G (IgG) level <500 mg/dL thereafter. Through May 2017, low-dose heparin infusion and ursodiol were given for veno-occlusive disease (VOD) prophylaxis (n = 36); after May 2017, ursodiol alone was used for VOD prophylaxis (n = 8). Patients aged <36 months who underwent transplant after December 2013 received a single dose of rituximab near discharge for prevention of autoimmune hemolytic anemia.
Cord-blood transplantation and DLI
UCB grafts were infused over 45 to 60 minutes. After June 2012, UCB washing post-thaw was not performed for patients weighing >10 kg to minimize cell loss. UCB units were thawed and diluted to 100 mL with cold dextran/albumin. An aliquot of 5 mL was then re-cryopreserved for possible DLI. For washed UCB grafts, the supernatant was pelleted, then recryopreserved. When indicated, DLI aliquots were diluted to 30 mL and administered after informed consent.
Posttransplant assessments
Chimerism was performed by the short tandem repeat method in whole blood and CD3+ fractions while peripheral blood lymphocyte reconstitution was monitored by flow cytometry. Immune reconstitution was assessed before conditioning and monitored posttransplant and was compared with the lower limit of age-appropriate normal range for CD3, CD4, CD8, and CD4/CD45RA/CD62L+ (surrogate recent thymic emigrants) and natural killer cell numbers.17 Neurodevelopmental assessments posttransplant were performed as previously described.1
Research immune studies
T-cell receptor (TCR) excision circles (TREC) was measured by using real-time polymerase chain reaction as previously described,18 with minor modifications. TCR repertoire diversity was determined by quantifying TCRβ CDR3 size variability via spectratyping as described elsewhere.19 Normal TCRβ CDR3 size was characterized as a Gaussian distribution, containing 8 to 10 peaks for each Vβ subfamily. Plasma samples (day 0) were frozen at −80°C until alemtuzumab measurement.20
Statistical methods
Using GraphPad Prism 7 software (San Diego, CA), continuous variables were analyzed by using the Mann-Whitney U test, and categorical variables were analyzed by using Fisher's exact test or χ2 analysis. Survival and cumulative incidence curves were calculated by using the noncompeting risk Kaplan-Meier method and compared by using the log-rank (Mantel-Cox) test. For purposes of calculating event-free survival (EFS), events were defined as death or graft failure.
Results
Patient characteristics
Patient demographic characteristics are shown in Table 1. The median age was 1.7 years. Sixty-one percent of patients were male, and 66% were white. Most patients (68%) had newly diagnosed leukodystrophies or other IEM. All but 1 patient with IEM exhibited progressive neurodevelopmental deterioration before transplantation. Sixteen (36%) patients were CMV seropositive; however, 5 patients were receiving supplemental IgG before transplantation, and an additional 6 were <12 months of age at time of transplantation, when maternal CMV antibodies may be detectable.21,22 The median length of stay was 43 days (range, 27-148 days).
Graft characteristics
The infused, post-thaw median total nucleated cell and CD34+ cell doses were 9.1 × 107/kg (range, 2.3-24.8 × 107/kg) and 3.31 × 105/kg (range, 0.92-9.24 × 105/kg), respectively. Most UCB grafts were HLA-mismatched, and more than one-half were mismatched for sex and/or ABO blood type (Table 1).
Neutrophil and platelet engraftment
All 44 patients engrafted with donor neutrophils at a median of 15 days posttransplant (range, 10-33) (Figure 1A). One patient experienced secondary graft failure on day 38 post-UCBT and underwent a successful second UCBT within 3 months using busulfan-based reduced toxicity conditioning. In 36 evaluable patients, median platelet engraftment ≥20 000/µL was at 32 days posttransplant (range, 15-56 days) (Figure 1B). Eight patients were unevaluable for platelet engraftment; seven due to a higher platelet transfusion threshold (bleeding [n = 3], sickle cell disease [n = 3], platelet aggregation defect [n = 1]), and one due to secondary graft failure. In 42 evaluable patients, median platelet engraftment ≥50 000/µL occurred 37 days posttransplantation (range, 27-79 days). Two patients were unevaluable due to requiring higher thresholds (bleeding, n = 1; graft failure, n = 1). Donor-specific antibodies were tested in 31 patients and were negative in 30. The only patient with positive donor-specific antibodies remains fully donor at >3 years posttransplant (data not shown). Significantly, both patients with osteopetrosis and all patients with transfusion-dependent anemia engrafted, highlighting the efficacy of the same RIC regimen across all major NMD categories.
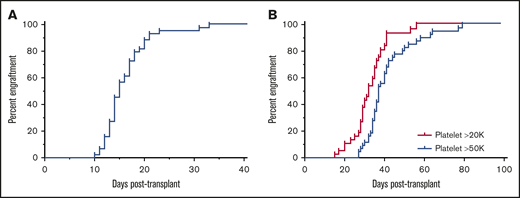
Neutrophil and platelet engraftment. (A) Neutrophil engraftment was defined as the first of 3 consecutive days with an absolute neutrophil count >500 cells/µL. Neutrophil engraftment occurred in all patients at a median of 15 days posttransplantation (range, 10-33 days). (B) Platelet engraftment was defined as the first of 7 consecutive days with a platelet count >20 000 cells/µL or >50 000 cells/µL without transfusion support. Platelet engraftment >20 000 cells/µL occurred in all evaluable patients at a median of 32 days posttransplantation (range, 15-56 days), and platelet engraftment >50 000 cells/µL occurred in all evaluable patients at a median of 37 days posttransplantation (range, 27-79 days).
Neutrophil and platelet engraftment. (A) Neutrophil engraftment was defined as the first of 3 consecutive days with an absolute neutrophil count >500 cells/µL. Neutrophil engraftment occurred in all patients at a median of 15 days posttransplantation (range, 10-33 days). (B) Platelet engraftment was defined as the first of 7 consecutive days with a platelet count >20 000 cells/µL or >50 000 cells/µL without transfusion support. Platelet engraftment >20 000 cells/µL occurred in all evaluable patients at a median of 32 days posttransplantation (range, 15-56 days), and platelet engraftment >50 000 cells/µL occurred in all evaluable patients at a median of 37 days posttransplantation (range, 27-79 days).
Donor chimerism
Whole-blood chimerism was ≥90% donor in 37 of 42 patients at 100 days, 33 of 38 patients at 180 days, 30 of 37 patients at 1 year, and 22 of 31 patients at 2 years posttransplantation. The number of patients with CD3 chimerism ≥90% donor were similar (23 of 31 patients at 100 days, 20 of 32 patients at 180 days, 27 of 35 patients at 1 year, and 22 of 28 patients at 2 years posttransplantation (Table 2; Figure 2). Among patients with mixed (<95% donor) whole-blood chimerism at 1 year, the median donor contribution to hematopoiesis was 85% (range, 47%-91%).
Complications posttransplantation (N = 44)
| Complication | Value |
|---|---|
| Engraftment syndrome, n (%) | 16 (36) |
| Acute GVHD, n (% of GVHD) | 21 (48) |
| Grade I | 9 (43) |
| Skin only | 9 (43) |
| Grade II | 7 (33) |
| Skin only | 6 (29) |
| Skin + gastrointestinal | 1 (5) |
| Grade III | 5 (24) |
| Skin + gastrointestinal | 2 (10) |
| Gastrointestinal only | 3 (14) |
| Grade IV | 0 (0) |
| GVHD grade I-IV onset, median (range), d | 47 (13-145) |
| GVHD grade II-IV onset, median (range), d | 39.5 (14-145) |
| Whole-blood chimerism (donor percentage) at 1 y (n = 37), n (%) | |
| >95% | 28 (76) |
| 90-95% | 2 (5) |
| 75-89% | 5 (14) |
| 50-74% | 1 (3) |
| 25-49% | 1 (3) |
| <25% | 0 (0) |
| T-cell chimerism (donor percentage) at 1 y (n = 35), n (%) | |
| >95% | 22 (63) |
| 90%-95% | 5 (14) |
| 75%-89% | 4 (11) |
| 50%-74% | 3 (9) |
| 25%-49% | 1 (3) |
| <25% | 0 (0) |
| Opportunistic infections at 1 y, n (% of infections) | 27 (61) |
| Adenovirus | 11 (41) |
| EBV | 10 (37) |
| HHV-6 | 9 (33) |
| CMV | 6 (22) |
| Mycobacteria | 1 (4) |
| Fungal | 1 (4) |
| Complication | Value |
|---|---|
| Engraftment syndrome, n (%) | 16 (36) |
| Acute GVHD, n (% of GVHD) | 21 (48) |
| Grade I | 9 (43) |
| Skin only | 9 (43) |
| Grade II | 7 (33) |
| Skin only | 6 (29) |
| Skin + gastrointestinal | 1 (5) |
| Grade III | 5 (24) |
| Skin + gastrointestinal | 2 (10) |
| Gastrointestinal only | 3 (14) |
| Grade IV | 0 (0) |
| GVHD grade I-IV onset, median (range), d | 47 (13-145) |
| GVHD grade II-IV onset, median (range), d | 39.5 (14-145) |
| Whole-blood chimerism (donor percentage) at 1 y (n = 37), n (%) | |
| >95% | 28 (76) |
| 90-95% | 2 (5) |
| 75-89% | 5 (14) |
| 50-74% | 1 (3) |
| 25-49% | 1 (3) |
| <25% | 0 (0) |
| T-cell chimerism (donor percentage) at 1 y (n = 35), n (%) | |
| >95% | 22 (63) |
| 90%-95% | 5 (14) |
| 75%-89% | 4 (11) |
| 50%-74% | 3 (9) |
| 25%-49% | 1 (3) |
| <25% | 0 (0) |
| Opportunistic infections at 1 y, n (% of infections) | 27 (61) |
| Adenovirus | 11 (41) |
| EBV | 10 (37) |
| HHV-6 | 9 (33) |
| CMV | 6 (22) |
| Mycobacteria | 1 (4) |
| Fungal | 1 (4) |
EBV, Epstein-Barr virus; HHV, human herpesvirus 6.

Donor chimerism analysis. Donor chimerism was measured by short tandem repeat analysis in peripheral whole blood and, when sufficient CD3+ cells were present in peripheral blood, in sorted CD3+ cells. Donor chimerism >95% in whole blood was seen in 81%, 83%, 76%, and 65% of patients and in 71%, 53%, 63%, and 71% for CD3+ cells at days (D) 100 and 180 and at 1 and 2 years posttransplantation, respectively.
Donor chimerism analysis. Donor chimerism was measured by short tandem repeat analysis in peripheral whole blood and, when sufficient CD3+ cells were present in peripheral blood, in sorted CD3+ cells. Donor chimerism >95% in whole blood was seen in 81%, 83%, 76%, and 65% of patients and in 71%, 53%, 63%, and 71% for CD3+ cells at days (D) 100 and 180 and at 1 and 2 years posttransplantation, respectively.
Transplant-related morbidity
No patients experienced transplantation-associated microangiopathy, interstitial pneumonitis syndrome, pericardial effusion requiring medical or surgical interventions, or VOD. In addition, no cases of macroscopic hematuria or hemorrhagic cystitis were observed. Autoimmune hemolytic anemia occurred in 10 patients, 7 of whom received an ABO mismatched graft; however, only 1 patient required systemic corticosteroids following prompt upfront rituximab therapy. Taken together, regimen-related toxicities were limited.
GVHD
Engraftment syndrome associated with noninfectious fever, rash, and capillary leak necessitating corticosteroids23,24 was reported in 7 subjects (16%) at a median of 11 days posttransplant (range, 8-13 days). The cumulative incidence of grades II to IV and III to IV acute GVHD by day 180 was 27% (95% confidence interval [CI], 10-48) and 11% (95% CI, 1-40), respectively (Figure 3A). The median day of grade II to IV acute GVHD onset was 40 days posttransplantation (range, 14-145 days). Skin was the most frequently involved organ (Table 2). The cumulative incidence of any chronic GVHD was 42% (95% CI, 25-58) (Figure 3B), and all were limited to mild skin rash controlled with topical steroid therapy. No patient developed severe/extensive chronic GVHD. Importantly, 72% of those who developed chronic GVHD did so during taper of immunosuppression and were easily controlled with topical therapy and slowing the taper or stepping back one dose level.

GVHD. (A) The cumulative incidence of grade II to IV GVHD was 27% (95% CI, 10-48) and the cumulative incidence of grade III to IV GVHD was 11% (95% CI, 1-40). The median onset day for grade II to IV GVHD was 40 days posttransplantation (range, 14-145 days). (B) The cumulative incidence of limited chronic GVHD was 42% (95% CI, 25-58). No patients developed severe or extensive chronic GVHD.
GVHD. (A) The cumulative incidence of grade II to IV GVHD was 27% (95% CI, 10-48) and the cumulative incidence of grade III to IV GVHD was 11% (95% CI, 1-40). The median onset day for grade II to IV GVHD was 40 days posttransplantation (range, 14-145 days). (B) The cumulative incidence of limited chronic GVHD was 42% (95% CI, 25-58). No patients developed severe or extensive chronic GVHD.
Survival
The median follow-up of surviving patients is 49.5 months (range, 13.3-98.4 months). Overall survival at 1 and 5 years posttransplantation is 95% (95% CI, 83-99) and 85% (95% CI, 67-94), respectively. EFS at 1 and 5 years posttransplantation is 93% (95% CI, 80-98) and 83% (95% CI, 66-92) (Figure 4). Treatment-related mortality was 5% (95% CI, 0-46) at 1 year posttransplantation. Two patients died before 1 year, one from adenoviral/CMV/parainfluenza disease at day 121, and one from progression of Tay-Sachs disease complicated by adenoviral pancreatitis at day 185 posttransplantation. Three additional patients (neurotrophic Gaucher disease, n = 2; metachromatic leukodystrophy [MLD], n = 1) died at 15, 41, and 47 months posttransplantation. All three of these patients were significantly affected by their IEM before transplantation and died of complications of their pretransplantation comorbidities.

Survival. (A) Overall survival was 95% (95% CI, 83-99) at 1 year, and EFS was 93% (95% CI, 80-98) at 1 year. (B) Overall survival was 85% (95% CI, 67-94) at 5 years posttransplantation and 83% (95% CI, 66-92) at 5 years posttransplantation.
Survival. (A) Overall survival was 95% (95% CI, 83-99) at 1 year, and EFS was 93% (95% CI, 80-98) at 1 year. (B) Overall survival was 85% (95% CI, 67-94) at 5 years posttransplantation and 83% (95% CI, 66-92) at 5 years posttransplantation.
Enzyme activity and neurodevelopmental outcomes
In the subset of patients with IEM with enzyme activities measured, 16 of 16 at 100 days posttransplantation, 15 of 15 at 180 days posttransplantation, and 14 of 14 at 1 year posttransplantation had enzyme activities in the unaffected range. There were 4 patients with symptomatic infantile Krabbe disease, in stage 2 of disease progression.25 These patients stabilized at a low functional level and fared better than our historical disease stage–matched control subjects receiving myeloablative conditioning (supplemental Figure 2). The 2 patients with symptomatic late infantile Krabbe disease stabilized in the motor area and continue to gain cognitive skills. Two of the patients with symptomatic juvenile Krabbe disease are walking and attending regular school, and the third progressed and is functioning at a low level. Of the 2 patients with late-infantile onset symptomatic MLD, one is able to walk with a walker, and the other died in the peri-transplantation period of infection. Four of the patients with juvenile-onset MLD are able to walk, speak, and function in the delayed range but are stable. The patient with asymptomatic adrenoleukodystrophy is able to walk and talk despite his behavioral difficulties and developmental delay related to the additional diagnosis of autism. One patient with Hurler syndrome who underwent transplant at 22 months of age and 2 patients with Hunter syndrome who underwent transplant at 10 months and 1.25 years of age continue to gain skills but have mild global developmental delay.
Immune reconstitution
Natural killer and B lymphocytes achieved normal numbers by day 100 posttransplantation, whereas T-cell reconstitution was slower, accelerating between day 100 and 180 posttransplantation (Figure 5A; supplemental Figure 3). TREC and TCR Vβ repertoire were measured to better assess thymopoiesis. TCR Vβ repertoire was near pretransplantation values by 180 days posttransplantation, whereas TREC copies began to rise by 100 days posttransplantation, reaching normal values ∼270 days posttransplantation concordant with flow cytometry values for recent thymic emigrants (Figure 5B-C). Twenty-eight (74%) of the 38 engrafted and evaluable surviving patients were off systemic immunosuppression at 1 year posttransplantation, and 30 (79%) of 38 patients did not require IgG supplementation at 1 year posttransplantation.
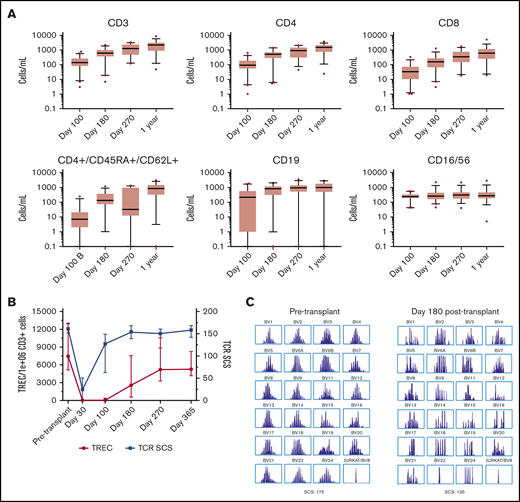
Immune reconstitution posttransplantation. (A) Absolute numbers of CD3, CD4, CD8+, and CD4/CD45RA/CD62L+ T cells, B cells, and natural killer cells as measured by flow cytometry are shown. Each box extends from the 25th to 75th percentile with a line at the median and whiskers extending 1.5 times the interquartile range. (B) The number of signal joint TRECs, normalized per 1 × 106 CD3 cells, and TCR Vβ repertoire, measured as TCR spectrotype complexity score (TCR SCS), which counts the number of peaks/TCR Vβ subfamily (maximum score, 208); they were measured pretransplantation and posttransplantation to better assess thymic recovery. (C) A representative example of pretransplantation and posttransplantation TCR SCS.
Immune reconstitution posttransplantation. (A) Absolute numbers of CD3, CD4, CD8+, and CD4/CD45RA/CD62L+ T cells, B cells, and natural killer cells as measured by flow cytometry are shown. Each box extends from the 25th to 75th percentile with a line at the median and whiskers extending 1.5 times the interquartile range. (B) The number of signal joint TRECs, normalized per 1 × 106 CD3 cells, and TCR Vβ repertoire, measured as TCR spectrotype complexity score (TCR SCS), which counts the number of peaks/TCR Vβ subfamily (maximum score, 208); they were measured pretransplantation and posttransplantation to better assess thymic recovery. (C) A representative example of pretransplantation and posttransplantation TCR SCS.
Infections
Following conditioning, the cumulative incidence of first opportunistic infection at 1 year posttransplantation was 61% (95% CI, 49 to 72) with a median onset day of 18 days posttransplantation (range, −27 to 224 days) (supplemental Figure 4; Table 2). Eight patients had infections with >1 organism. The majority of opportunistic infections were asymptomatic viremia. One patient developed Candida parapsilosis infection at day 24, and another was found to have asymptomatic pulmonary Mycobacterium kansasii infection on a screening chest radiograph at the 3-month visit. The majority of patients received appropriate pharmacologic therapy for viremia. The patient with congenital intestinal atresia developed Epstein-Barr virus–positive plasmacytoma 6 months after small bowel transplantation and ∼2 years post-UCBT; the patient is alive and in remission >5 years post-UCBT. Two patients developed refractory adenoviremia and were treated with virus-specific T cells on investigational new drug protocols and died.
Alemtuzumab levels
Most patients (n = 39) received a cumulative dose of 1.2 mg/kg of alemtuzumab. One patient received 0.7 mg/kg due to active CMV viremia and parainfluenza at the time of conditioning, whereas 4 patients (1 with hemophagocytic lymphohistiocytosis and 3 with hemoglobinopathies) received 2.2 mg/kg. Twenty-six patients had samples drawn on the day of transplantation for alemtuzumab levels. The median alemtuzumab level was 0.58 μg/mL (range, 0-2.82 μg/mL) (Figure 6A). Notably, patients exhibiting day 0 levels above the median had fewer circulating T cells at days 30 and 60 posttransplantation, exhibiting greater early lymphotoxicity with resolution by day 100 (Figure 6B-D). At 1 year posttransplantation, there was no significant difference in GVHD, infection, or mixed chimerism in the whole-blood or T-cell fractions between patients with high or low day 0 alemtuzumab levels (supplemental Figure 5).
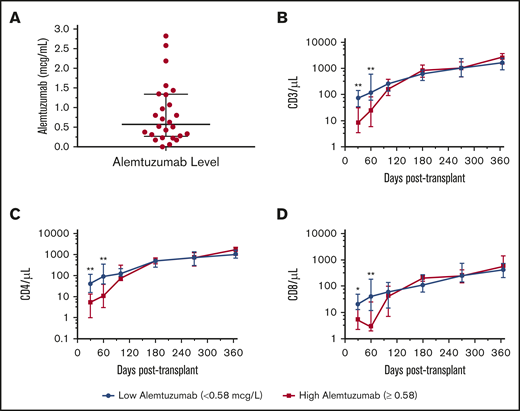
Alemtuzumab levels and immune reconstitution. (A) Day 0 alemtuzumab levels were measured in 26 subjects. The median alemtuzumab concentration was 0.58 μg/mL (range, 0-2.82 μg/mL) at day 0. (B-D) Subjects with alemtuzumab above the median had lower absolute CD3, CD4, and CD8 counts at 30 and 60 days posttransplantation compared with subjects with alemtuzumab levels below the median. *P < .05; **P < .01.
Alemtuzumab levels and immune reconstitution. (A) Day 0 alemtuzumab levels were measured in 26 subjects. The median alemtuzumab concentration was 0.58 μg/mL (range, 0-2.82 μg/mL) at day 0. (B-D) Subjects with alemtuzumab above the median had lower absolute CD3, CD4, and CD8 counts at 30 and 60 days posttransplantation compared with subjects with alemtuzumab levels below the median. *P < .05; **P < .01.
Cord-derived DLI
Bone marrow transplantation (BMT) physicians were offered 3 clinical criteria to use DLI: those with potentially life-threatening viremia or viral infections (n = 10), mixed T-cell chimerism (n = 9), or sluggish T-cell reconstitution (<100/µL after 2 months, n = 7). Seven of the 20 cDLI recipients had >1 indication for cDLI, most commonly infection with delayed CD3 recovery (n = 4). Twenty patients received cDLI at a median of 66 days posttransplantation (range, 14-124 days) to foster homeostatic T-cell expansion in the first 4 to 6 months until effective thymopoiesis, with the majority (n = 14) receiving DLI between days 38 and 75 (supplemental Figure 6A). The 2 patients who received cDLI before day 30 did so due to very-high-grade adenoviremia, and they represent the only patients who experienced transplant-related mortality, dying despite additional virus-specific third-party DLI. In fact, alemtuzumab dose de-escalation based on previous viral infection was introduced after these early back-to-back subjects. The median cell dose of the cDLI was 7.56 × 105 CD3/kg (range, 2.36-24.5 × 105 CD3/kg) with negligible CD34+ content (median, 1.58 × 104/kg; range, 0.79-5.04 × 104 CD34/kg).
Viability post-thaw was excellent, with a median viability of 94% (range, 76%-100%). Improvement in donor chimerism was seen in 5 of 9 patients, reduction in viral load and/or clinical symptoms of viral infection in 6 of 10 patients, and absolute CD3 counts rose in 7 of 7 patients (Figure 7). The kinetics of CD3, CD4, and CD8 cell reconstitution for subjects who received cDLI between days 38 and 75 (n = 14) were examined between 2 and 12 months after UCBT and compared with results in those who never received cDLI (n = 24) (supplemental Figure 6B-D). Although the patients who received cDLI exhibited an increase in the rate of T-cell reconstitution, the increase did not reach statistical significance, reflecting the small cohorts and 3 different indications. Eight patients who received cDLI developed grade I to II skin GVHD at a median of 19 days post-cDLI (range, 4-81 days). Two patients with a history of acute GVHD received cDLI without GVHD flare. No patients who received cDLI developed grade III or IV acute GVHD or severe/extensive chronic GVHD. There was no difference in the cumulative incidence of acute GVHD between those who received cDLI and those who did not (47% [95% CI, 23-69] and 48% [95% CI, 28-66], respectively; P = .6) (Figure 7D).

Effect of cDLI posttransplantation. (A) Nine subjects received cDLI for mixed whole-blood or T-cell chimerism. Improvement in T-cell chimerism was reported in the majority of patients at 120 days after cDLI. (B) Ten subjects received cDLI for viral infection (7 with viremia, 3 with symptomatic viral gastroenteritis). Improvement in viremia and/or clinical symptoms was seen in 6 of 10 subjects. (C) Seven subjects received cDLI for delayed immune reconstitution. Improvement in absolute CD3 count was noted in all 7 subjects. (D) The cumulative incidence of acute GVHD or progression of acute GVHD was 47% (95% CI, 23-69) in subjects who received cDLI and 48% (95% CI, 28-66) in those who did not.
Effect of cDLI posttransplantation. (A) Nine subjects received cDLI for mixed whole-blood or T-cell chimerism. Improvement in T-cell chimerism was reported in the majority of patients at 120 days after cDLI. (B) Ten subjects received cDLI for viral infection (7 with viremia, 3 with symptomatic viral gastroenteritis). Improvement in viremia and/or clinical symptoms was seen in 6 of 10 subjects. (C) Seven subjects received cDLI for delayed immune reconstitution. Improvement in absolute CD3 count was noted in all 7 subjects. (D) The cumulative incidence of acute GVHD or progression of acute GVHD was 47% (95% CI, 23-69) in subjects who received cDLI and 48% (95% CI, 28-66) in those who did not.
Discussion
Here we report the outcomes of the largest prospective cord blood transplant trial for NMDs. We formally tested the efficacy and safety of a biologically rational modification of the classic “Campath-Flu-Mel” RIC regimen using intermediate-timed alemtuzumab aided by the addition of hydroxyurea and thiotepa. Although previous RIC regimens have been plagued by graft failure, this regimen was found to be very effective in permitting successful engraftment for single-unit UCB grafts in a broad range of disorders encompassing all major categories of NMDs. Unlike patients with leukemia, the current study patients were chemotherapy naive and thus at increased risk for rejection due to a robust bone marrow environment, and the vast majority had intact immunity with respect to graft rejection.
There are several reasons this "Pitt-RIC" regimen could be transformative for children afflicted with a wide variety of NMDs. First, the graft failure rate of <5% seen in this trial is the lowest ever reported for unmanipulated cord blood grafts in malignant or nonmalignant diseases, even when MAC regimens are included. Notably, in recent systematic analyses, graft failure rates of 8% to 15% have been reported,26-29 consistent with older series.30 The remarkably early neutrophil engraftment at a median of 15 days likely reflects the marrow stroma sparing by our RIC regimen. Notably, engraftment was attainable even for patients with osteopetrosis, 3 different forms of transfusion-dependent anemia, and immune dysregulation syndromes, in which engraftment has been challenging even in MAC regimens utilizing bone marrow grafts. We used hydroxyurea to induce mild neutropenia and capitalize on the associated increase in stem cells entering cell cycle.31 This approach was intended to increase the impact of the alkylator drugs on host stem cells even at reduced doses. Efficient depletion of host hematopoietic reserves was further aided by the addition of thiotepa to the regimen. Furthermore, cell loss was minimized as the UCB grafts were thawed, diluted, and infused without additional wash as described,32 contrasting with common pediatric practice. Significantly, the cumulative immuno-ablation afforded by the optimized alemtuzumab dosing paired with hydroxyurea and the addition of thiotepa resulted in only 1 patient experiencing secondary graft failure. The reemergence of recipient-derived hematopoiesis and resolution of neutropenia by day 42 posttransplantation in the 1 patient who experienced graft failure shows that this regimen fulfills the tenets of reduced intensity, mitigating the risks associated with prolonged or persistent pancytopenia. At the time of hospital discharge, patients were transfusion independent, and most patients maintained full (≥95%) donor chimerism at 2 years posttransplantation with none <25%, comparing favorably to other RIC regimens for NMDs.33-36 In addition, in the patients with IEM, leukocyte-based enzyme levels were within normal range with up to 2 years follow-up.
Second, this regimen was associated with a very low 1-year TRM of 5%, resulting in >90% EFS at 1 year. Although there are few prospective trials of cord blood grafts for IEM, systematic reviews have reported 12% to 28% TRM at 1 year.27,37 Contemporary reports of pediatric cohorts utilizing bone marrow or mobilized peripheral blood stem cell grafts have reported TRM of 12% to 15% at 1 year,38 whereas mortality and graft failure with high-dose T-cell–depleted peripheral blood stem cell transplantation for immune deficiencies was 15% and 12%, respectively, at 1 year.39 Although mild to moderate mucositis was uniform, no patient developed VOD, hemorrhagic cystitis, idiopathic pneumonia syndrome, pericardial effusion requiring medical or surgical intervention, or renal replacement therapy. In addition, no patient experienced transplant-associated microangiopathy. The use of split-dose melphalan rather than the single dose used in our trial may have favorably affected the low TRM, resulting in minimal regimen-related toxicity or mortality. Engraftment syndrome was low, with no apparent association with GVHD risk. Despite 43 of 44 referred patients exhibiting symptoms or complications of their underlying disease, an extremely high survival rate was achieved. Significantly, more than one-half of our patients had progressive leukodystrophies, a patient population for whom mortality after myeloablation with busulfan-containing regimens exceeds 40% by 3 years post-UCBT, making these patients ineligible for MAC transplantation1,27,30 and leading to reluctance to refer for transplantation at many centers. This poor outcome is in sharp contrast to the current protocol, in which the 3-year EFS was 94% in the leukodystrophy subcohort and those patients with symptomatic infantile Krabbe disease stabilized at a low functional level that was better than that of the historical control subjects (supplemental Figure 2).
Third, a single-unit cord blood graft could be identified for all 44 eligible children regardless of ethnicity, showing the unique and unparalleled suitability of cord blood grafts even with HLA allele mismatches.12 More than one-half of our patients had at least 2 allele mismatches, yet neither engraftment, GVHD, nor survival were influenced by the degree of HLA mismatch. The excellent EFS following this RIC trial, often utilizing HLA-mismatched single cord blood, exceeds that reported in young children receiving bone marrow grafts from predominantly HLA-matched donors using alemtuzumab, fludarabine, and melphalan without the addition of hydroxyurea and thiotepa.40,41 The immunoablative combination of alemtuzumab, melphalan, and thiotepa may explain the clinically neutral nature of HLA mismatches on rejection, whereas the in vivo depletion of infused cord blood T cells by lingering day 0 alemtuzumab levels may explain the relatively low rates of GVHD. Grade III to IV GVHD occurred in only 10% of patients, in line with other intermediate-timing alemtuzumab regimens.35,42 Critical to quality of life, no patient developed severe/extensive chronic GVHD. Immune reconstitution also benefited in a clinically relevant way from lower cumulative doses of alemtuzumab.15 Although low-grade viremia was reported in 61% of patients at 1 year posttransplantation, this outcome is similar to those of other alemtuzumab-containing conditioning regimens in patients with nonmalignant disease, in which the incidence of viremia ranged between 50% and 81%.11,15,20,43 Importantly, only 2 patients in our study developed viral-related mortality, reflecting the favorable immune reconstitution and comparable to what is seen in antithymocyte globulin–containing conditioning regimens.38,44
An additional novel, although somewhat preliminary, finding is that T-cell reconstitution after pretransplant serotherapy can be supported by infusing a fraction of the original cord blood unit previously set aside and re-cryopreserved. Formal evaluation in a larger study is required to further characterize the merits of unmanipulated cDLI in the context of single-unit cord blood transplantation for mixed chimerism or refractory viral infections when pretransplant serotherapy drugs decline below lytic levels.
Lastly, and perhaps most importantly, this regimen is adaptable to less resource-rich countries because there is no need for real-time dose adjustment of any chemotherapy or serotherapy agent based on pharmacokinetic levels. Interestingly, not even pharmacokinetic dosing of busulfan, antithymocyte globulin, or fludarabine in a myeloablative setting has generated lower TRM and better EFS in retrospective analyses compared with our report.45,46 Widespread applicability of our regimen is supported by a uniquely enhanced safety profile as no patient required the use of expensive and potentially risky drugs such as defibrotide or eculizumab to overcome regimen-related toxicity.
In this relatively young population, there is also no clinically pressing need for complex or inherently expensive cell-expansion methodologies or adjunct supportive grafts such as double-cord or haplo-cord, highlighting the cost-effective and widely applicable benefits of an unmanipulated single-unit UCB graft for children with NMD.47-50
In summary, this article describes a highly effective and biologically rational RIC regimen applicable for a diverse range of inheritable NMDs with unrelated UCB grafts identifiable for all in whom HSCT is indicated. This novel chemotherapy-based regimen paired with simple alemtuzumab dosing strata prevented graft failure and aided immune reconstitution. The favorable outcome described may serve as a toxicity and efficacy reference for emerging gene therapy strategies as well. Although this is a single-center dataset, all patients had at least 12 months follow-up, and it is the largest prospective clinical trial to report on cord blood transplantation for NMDs.
Deidentified individual participant data and the study protocol that underlie the reported results will be made available 3 months after publication for a period of 5 years following the publication date to researchers who submit a methodologically sound proposal. Proposals for access should be sent to paul.szabolcs@chp.edu.
Acknowledgments
Most importantly, the authors thank the patients and their families. The study team thanks the BMT coordinators, Program For The Study of Neurodevelopment in Rare Disorders, The Center for Rare Disease Therapy, BMT nursing and clinical staff, BMT regulatory team, and the HLA and stem cell laboratories and staff at UPMC Children’s Hospital of Pittsburgh. The authors also thank Cincinnati Children’s Hospital Medical Center for alemtuzumab levels.
Immune reconstitution studies were supported by a grant from the National Institutes of Health, National Heart, Lung, and Blood Institute (R01HL091749) (P.S.).
Authorship
Contribution: P.S. conceived of the study; M.T.V.L., E.O.S., and P.S. wrote the clinical protocol; M.T.V.L., M.L.E., B.A.C., J.L.B., R.M.W., M.P., H.S., and P.S. collected clinical data; X.C. and M.J.H. performed immune reconstitution studies; R.A.M. measured the alemtuzumab levels; M.T.V.L. and P.S. codrafted the first version of the article; and all authors reviewed, edited, and approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for M.T.V.L. is Pediatric Blood and Marrow Transplant Program, University of Michigan, Ann Arbor, MI.
Correspondence: Paul Szabolcs, Division of Blood and Marrow Transplantation and Cellular Therapies, UPMC Children’s Hospital of Pittsburgh, 4401 Penn Ave, Pittsburgh, PA 15224; e-mail: paul.szabolcs@chp.edu.

