

Key Points
Juvenile xanthogranuloma (JXG) usually presents with lesions isolated to the skin; however, aggressive, disseminated forms also occur.
Identification of a novel MRC1-PDGFRB fusion in a child with JXG guided targeted therapy with dasatinib, leading to a dramatic response.
Introduction
Juvenile xanthogranuloma (JXG) family of lesions usually present as a localized nodular lesion confined to the dermis or hypodermis and collectively belong to the rare non-Langerhans cell histiocytic (LCH) group.1,2 Less often, JXG occurs as ≥1 aggressive extracutaneous lesions with potential for visceral involvement and organ dysfunction.3,4 Although localized JXG typically resolves spontaneously, progressive or systemic JXG can be life-threatening, requiring treatment with chemotherapy regimens similar to those used for LCH.2,5
Recent genomic investigations have identified the presence of kinase gene mutations in the majority of histiocytic tumors, including JXG.6-8 This new molecular information supports the investigation of targeted therapies to treat histiocytic neoplasms, particularly those that do not respond to frontline therapy.
Case description
A 4-month-old female infant presented with an enlarging mass lateral to the left nipple. Computed tomography (CT) scanning revealed a large left-chest-wall mass, measuring 5.0 × 2.9 × 2.6 cm, eroding the left fourth and fifth ribs and extending into subcutaneous fat (Figure 1A,C). Multiple areas of pleural thickening were noted over the left hemithorax, with the largest measuring 1.4 × 1.8 cm (Figure 1B). Left hilar and paracardiac lymphadenopathy was observed, with the largest lymph node measuring 0.8 × 0.9 cm. No pulmonary parenchymal lesions were noted. A staging positron emission tomography (PET) scan showed markedly increased uptake of fluorodeoxyglucose (FDG) in the left-chest-wall mass with areas of pleural thickening exhibiting only slightly increased metabolic activity. The PET-CT scan and a skeletal survey revealed no additional abnormalities. Initial laboratory studies, including a peripheral blood count and differential, were normal, and a bone marrow biopsy and aspirate showed normocellular marrow with balanced and orderly trilineage hematopoiesis.
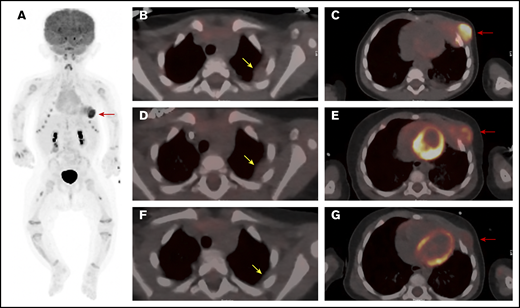
PET/CT scan of the chest at 3 time points. (A) Whole body anterior projection image from FDG PET/CT scan at diagnosis. Red arrow points to the FDG-avid left-sided chest wall mass. Sites of soft tissue uptake in both thighs represent injection sites from recent immunizations. Transverse fusion PET/CT sections through the thorax at baseline (B-C), following upfront standard therapy with vinblastine and steroids (D-E), and after therapy with dasatinib (F-G). At baseline, the mass extends into the subcutaneous fat of the left chest wall anteriorly and abuts the pericardium (C). The response to standard therapy was suboptimal with only mild reduction in size and FDG uptake (D-E). (G) Marked reduction in size as well as FDG avidity in response to dasatinib therapy. Yellow arrows point to sites of pleural thickening that were not FDG avid (1.4 × 1.8 cm). Note that the appearance of the pleural thickening did not change after standard therapy, but nearly resolved following dasatinib. Red arrows point at the large left-chest-wall mass centered at the left anterior fourth and fifth ribs and causing rib destruction.
PET/CT scan of the chest at 3 time points. (A) Whole body anterior projection image from FDG PET/CT scan at diagnosis. Red arrow points to the FDG-avid left-sided chest wall mass. Sites of soft tissue uptake in both thighs represent injection sites from recent immunizations. Transverse fusion PET/CT sections through the thorax at baseline (B-C), following upfront standard therapy with vinblastine and steroids (D-E), and after therapy with dasatinib (F-G). At baseline, the mass extends into the subcutaneous fat of the left chest wall anteriorly and abuts the pericardium (C). The response to standard therapy was suboptimal with only mild reduction in size and FDG uptake (D-E). (G) Marked reduction in size as well as FDG avidity in response to dasatinib therapy. Yellow arrows point to sites of pleural thickening that were not FDG avid (1.4 × 1.8 cm). Note that the appearance of the pleural thickening did not change after standard therapy, but nearly resolved following dasatinib. Red arrows point at the large left-chest-wall mass centered at the left anterior fourth and fifth ribs and causing rib destruction.
The ultrasound-guided biopsy revealed a dense, bland histiocytic infiltrate in a background of skeletal muscle (Figure 2A-F). Immunohistochemical (IHC) staining demonstrated positivity for CD163, CD14, factor XIIIa, fascin, and CD33, and partial light positivity for CD45, CD43, and S100. Histiocytic cells showed nonspecific blush cytoplasmic staining, equivocal positivity for CD1a and CD117 and negativity for BRAF V600E (VE1), langerin, ALK, AFB, CD34, CD21, synaptophysin, cytokeratin AE1/AE3, TFE-3, and desmin. Based on pathology and clinical findings, a diagnosis of JXG was made. Molecular testing was negative for the BRAF V600E mutation; however, whole-genome and RNA-sequencing analyses revealed a novel somatic t(5;10)(q32; p12.33) translocation resulting in the formation of an in-frame MRC1-PDGFRB gene fusion (Figure 2G). IHC staining, performed to assess activation of the platelet-derived growth factor receptor (PDGFR) β (PDGFRB) pathway, revealed diffuse expression of cyclin D1 in tumor cells (Figure 2F).
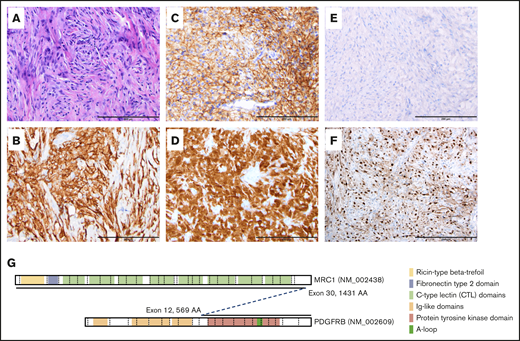
Histologic and immunohistochemical studies and schematic representation of somatic translocation. (A) Hematoxylin-and-eosin–stained section, original magnification ×20, showing diffuse sheets of histiocytoid cells set in a fibroinflammatory background. Immunohistochemical staining shows diffuse positivity for CD163 (B), CD33 (C), and factor XIIIa (D). No staining is noted for BRAF VE1 (E) and cyclin D1 (F) stains diffusely positive. (A-F) Scale bars, 200 μm. (G) Schematic representation of the MRC1-PDGFRB translocation.
Histologic and immunohistochemical studies and schematic representation of somatic translocation. (A) Hematoxylin-and-eosin–stained section, original magnification ×20, showing diffuse sheets of histiocytoid cells set in a fibroinflammatory background. Immunohistochemical staining shows diffuse positivity for CD163 (B), CD33 (C), and factor XIIIa (D). No staining is noted for BRAF VE1 (E) and cyclin D1 (F) stains diffusely positive. (A-F) Scale bars, 200 μm. (G) Schematic representation of the MRC1-PDGFRB translocation.
Once the diagnosis was established, the patient began treatment with vinblastine (0.2 mg/kg IV weekly) and prednisolone (13.3 mg/m2 orally thrice daily). The chest wall mass showed a subjective decrease in size upon initiation of therapy but regrew quickly thereafter. After 6 weeks, the mass was unchanged in size from the initial presentation, with only a slight reduction in FDG avidity (Figure 1D-E). Due to the inadequate clinical response, therapy was changed to single-agent dasatinib (80 mg/m2 per day) based on tumor positivity for the MRC1-PDGFRB fusion, which is predicted to lead to activation of the PDGFRB. Several recent reports support the use of imatinib or dasatinib in the treatment of neoplasms harboring PDGFRB-activating mutations.9,10
Following initiation of dasatinib therapy, the patient showed a steady and dramatic clinical response with a reduction in the size of the primary tumor. After 5 months of targeted therapy, PET-CT scanning showed a marked reduction in the size and activity of the primary tumor and near complete resolution of the pleural mass (Figure 1F-G). After 9 months of therapy, physical examination demonstrated continued improvement with the palpable mass measuring ∼1 cm in size.
Methods
Approval for this research was obtained from the Institutional Review Board of St. Jude Children’s Research Hospital.
Histology and immunohistochemical staining
Formalin-fixed paraffin-embedded tumor sections were cut at 4-μm thickness and placed on charged slides. Hematoxylin-and-eosin–stained sections were produced on a Ventana HE600 instrument. IHC-stained sections were produced using the Roche Ventana Benchmark Ultra and Dako Omnis systems and the following antibodies and dilutions: BRAF V600E (ready to use; Ventana Medical), CD33 (1/200; Leica Microsystems), CD163 (1/50; Cell Marque), cyclin D1 (clone SP4, ready to use; Cell Marque), and Factor XIIIa (1/40; Leica Microsystems).
Genomic testing
DNA and RNA were isolated from formalin-fixed paraffin-embedded tumor tissue and DNA isolated from a paired normal blood sample, according to standard protocols. DNA and RNA were subjected to library construction for whole-exome sequencing (WES) or RNA sequencing using the Illumina TruSeq Exome Enrichment Kit and the Illumina TruSeq Stranded Total RNA LT Kit, respectively. All libraries were sequenced using a paired end 2- × 125-bp cycle protocol on the Illumina NovaSeq. Sequence alignment was performed followed by identification and classification of tumor variants, as previously described.11
Results and discussion
Since the identification of BRAF V600E as the causal somatic genetic abnormality in LCH lesions, our understanding of the molecular pathogenesis of diverse histiocytic neoplasms has increased tremendously.6,8,12,13 This knowledge has significantly changed how these tumors are classified, monitored, and treated. Importantly, the incorporation of somatic genetic information into clinical care has informed the use of targeted therapies that are proving much more effective than conventional chemotherapy at ameliorating disease, particularly in patients with refractory or relapsed tumors.14-16
From a genetic perspective, the histiocytic disorders are generally considered “quiet” in terms of their genomic landscape, with most lesions showing only a single somatic alteration. By and large, these somatic alterations affect the RAS-MAPK pathway, with the frequency of specific genetic defects varying between disorders. For example, although the BRAF V600E mutation is identified in approximately one-half of all cases of LCH and Erdheim-Chester disease,7,17 it is rare in Rosai-Dorfman disease (RDD)18,19 and even rarer in JXG.20,21 An increasing number of studies in the literature describe the somatic mutations other than BRAF V600E found in JXG tumors. In a recent study including 55 patients with JXG, WES, transcriptome, targeted DNA, and/or targeted RNA sequencing identified kinase driver mutations in MAP2K1, NRAS, KRAS, CSF1R, and KIT, among others.8 Gene fusions involving NTRK1, BRAF, RET, and ALK were also identified. Additional reports have identified NF1, MAPK1, PI3KD and other mutations.6,20,22
To our knowledge, this case represents the first description of a patient with a JXG tumor harboring an MRC1-PDGFRB fusion. This fusion juxtaposes the intact tyrosine kinase domain of PDGFRB next to the extracellular domain of the c-type mannose receptor 1 (macrophage mannose receptor, MRC1, CD206) and is predicted to activate PDGFR signaling. Because MRC1 is expressed in macrophages and immature or dermal dendritic cells,23,24 it is very likely that the MRC1-PDGFRB fusion protein enhances PDGFR signaling output, thereby promoting JXG formation. PDGF induces expression of cyclin D1 via PDGFRB and the MAPK pathway.25,26 As WES identified no additional PDFGR/MAPK pathway mutations, the diffuse expression of cyclin D1 in the tumor cells is likely secondary to the MRC1-PDGFRB gene fusion. Further supporting a causal role for enhanced PDGFR signaling in histiocytosis, previous reports describe increased PRGFRA and PDGFRB expression in Erdheim-Chester disease, LCH, and RDD samples.27-29 In these studies, no comprehensive molecular studies of PDGFRA/B were performed. Therefore, it is not possible to know whether any activating mutations were present.
Preclinical studies and case reports of Philadelphia-like acute lymphoblastic leukemia harboring PDGFRB translocations have shown that these activated PDGFRB+ cells are responsive to treatment with kinase inhibitors such as dasatinib.10,30,31 Similarly, Utikal et al29 describe a patient with progressive skin and hepatic RDD, which stained positive for PDFGRB and KIT expression, whose disease completely resolved after 6 weeks of imatinib therapy. Based on these findings, we chose targeted therapy with dasatinib for the management of our patient and observed that this treatment produced a dramatic clinical response.
One weakness of this report is the short duration of treatment. Although BRAF and MEK inhibition generate rapid, complete response in histiocytic neoplasms, published reports demonstrate that this targeted therapy is usually not curative.14-16 Similarly, dasatinib treatment of chronic myeloid leukemia leads to high rates of remission but often cannot be stopped without relapse and is frequently continued indefinitely. For this patient, the initial planned treatment is for 1 year of dasatinib therapy. Evaluation at that time will determine ongoing management, including continuation of dasatinib with frequent reassessment of response and toxicity, discontinuation of therapy if no active disease or significant toxicity, or resection of the residual tumor if surgically accessible.
Overall, these data highlight the importance of completing genomic profiling of histiocytic lesions, including JXG, in order to identify somatic genetic alterations that can guide the use of targeted agents, particularly for patients whose disease fails to respond to conventional therapy.
Data-sharing requests may be e-mailed to the corresponding author, Patrick K. Campbell, at patrick.campbell@stjude.org.
Acknowledgments
The authors thank Jamie Maciaszek for careful review of this manuscript. The authors also thank the patient and her parents for their willingness to share this case report.
Authorship
Contribution: S.S.E., K.E.N., and P.K.C. collected data and wrote the manuscript; M.R.C., T.S., G.W., L.W., B.L.S., and J.P. interpreted data and created figures; and all authors reviewed and edited the manuscript.
Conflict-of-interest disclosure: K.E.N. receives research funding from Alpine Immune Sciences and Incyte Corporation. The remaining authors declare no competing financial interests.
Correspondence: Patrick K. Campbell, St. Jude Children’s Research Hospital, 262 Danny Thomas Pl, MS 514, Memphis, TN 38105; e-mail: patrick.campbell@stjude.org.

