

Key Points
High-density lipoprotein and apolipoprotein A-I enhance activated protein C cytoprotective activity.
High-density lipoprotein and apolipoprotein A-I significantly increase the rate at which activated protein C degrades cytotoxic extracellular histones.
Introduction
Activated protein C (APC) is a crucial regulator of blood clot formation, which it achieves by proteolytic degradation of procoagulant-activated cofactors factor V and factor VIII.1 Separately, APC can initiate cell signaling in numerous cell types to enhance cytoprotective responses to stress or toxic agents.2-4 APC cytoprotective activity on endothelial cells is mediated by agonism of protease-activated receptors (PARs), in particular PAR1 and PAR3, which it activates via proteolysis of defined extracellular cleavage sites.1,5,6 APC cannot efficiently cleave PARs unless already bound to other membrane receptors such as endothelial cell protein C receptor (EPCR),6 Mac-1,7 or apolipoprotein E receptor 2.8 Moreover, APC anti-inflammatory activity is at least in part related to its ability to neutralize cytotoxic extracellular histones released by dying tissue and neutrophil extracellular traps.9 Nevertheless, the molecular basis of how APC facilitates histone proteolysis to limit vascular injury is not well understood.
Despite significant evidence implicating the importance of PAR signaling and extracellular histone proteolysis in mediating the therapeutic benefits of APC,10-12 several aspects of how APC confers protection remain unresolved. In particular, APC is a relatively poor PAR1 agonist and is unlikely to be generated in vivo at the APC concentrations required to induce PAR1 signaling or extracellular histone proteolysis in vitro.9,13 Consequently, we sought to investigate whether endogenous blood-borne factors may play a role in regulating PAR1 signaling and extracellular histone proteolysis by APC.
Methods
Materials
Recombinant human APC was generated and characterized as previously described.13 Human thrombin was purchased from Haematologic Technologies Inc. Human plasma–purified high-density lipoprotein (HDL; >95% pure, #LP3-5MG), apolipoprotein A-I (Apo A-I; >95% pure, #ALP10-M), and Apo A-II (>95% pure, #A0792) were purchased from Merck-Sigma. HDL was isolated by sequential flotation ultracentrifugation and was composed of 55% to 45% lipid and 45% to 55% protein. HDL was refrigerated and used fresh, as loss of activity was observed when frozen or after prolonged storage, in keeping with previous reports.14 Escherichia coli–expressed recombinant Apo A-I (>95% pure, #SRP4693) was purchased from Merck-Sigma. Recombinant histone H3.1 was purchased from New England Biolabs, and antihistone H3.1 antibody (5192S) was purchased from Cell Signaling Technology. Phospholipid MP-Reagent (TS60.00) was purchased from Thrombinoscope. Anti-EPCR antibody RCR-252 and PAR1 antagonist (SCH530348) were purchased from Sigma and R&D Systems, respectively. The S1P1 receptor antagonist (Ex 26) was purchased from Bio-Techne.
Assessment of endothelial cell barrier permeability
Endothelial cell barrier permeability was determined as previously described.13,15 Briefly, EA.hy926 cells (ATCC) or human umbilical vein endothelial cells (PromoCell) were grown to confluence on polycarbonate membrane transwells (Merck) and incubated with APC and other reagents as indicated. Antibodies or antagonists when used were incubated with cell monolayers for 30 minutes before addition of APC. After 3 hours, cells were treated with thrombin (5 nM) for 10 minutes, after which the cells were washed and incubated with 0.67 mg/mL Evans Blue with 4% bovine serum albumin (Sigma). Changes in endothelial cell barrier permeability were then determined as previously described.15
Visualization of histone proteolysis by APC
Recombinant histone H3.1 (New England Biolabs) was incubated with APC in the presence or absence of Apo A-I or HDL at stated concentrations for 2 hours at 37°C. Histone proteolysis was assessed by immunoblotting using an antihistone H3.1 antibody (5192S) that detects remaining intact or cleaved histone H3.1.
Results and discussion
HDL is an abundant plasma lipoprotein that has previously been reported to enhance APC anticoagulant activity via acceleration of APC-mediated factor Va proteolysis.16 To determine whether HDL can also modulate non-anticoagulant APC functions such as protection of endothelial barrier integrity in response to thrombin-induced hyperpermeability, endothelial cells were exposed to HDL, and its ability to mediate APC-mediated protection of the endothelial cell barrier integrity was assessed. Interestingly, HDL, but not low-density lipoprotein (LDL), markedly enhanced APC barrier-protective function, such that half-maximal barrier protection occurred at five- to 10-fold lower APC concentrations compared with when HDL was absent (Figure 1A-B). Importantly, HDL alone exhibited negligible ability to limit endothelial barrier permeability in the absence of APC. HDL titration revealed that half-maximal enhancement of APC barrier function was achieved at ∼0.5 mg/mL HDL (Figure 1C), which is within the normal concentration range of plasma HDL (0.4-0.6 mg/mL). These data highlight a potential role for HDL in the protection of inflamed vasculature via enhancement of APC signaling.
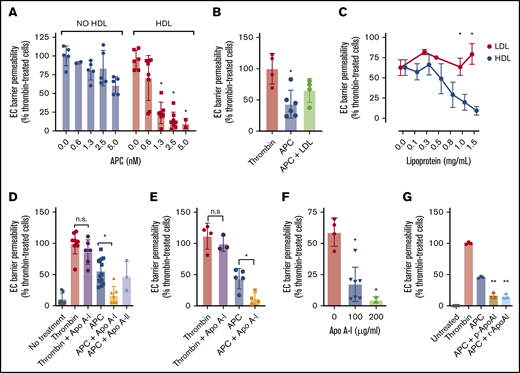
HDL and Apo A-I both enhance endothelial cell (EC) barrier protection by APC against thrombin-induced permeability. (A) The ability of the plasma lipoprotein HDL (1 mg/mL; red) to modulate protection against thrombin (5 nM) disruption of the EC barrier by APC (0.6-5 nM; blue) was measured. (B) Similarly, the ability of LDL (1 mg/mL) to mediate enhancement of APC barrier protection was tested under the same experimental conditions. (C) Evaluation of HDL and LDL enhancement of APC-mediated barrier protection was performed by titration of either HDL (blue) or LDL (red) in the presence of APC (5 nM) before thrombin (5 nM) exposure. (D) The ability of Apo A-I or Apo A-II (both 200 µg/mL) to enhance APC-mediated barrier protection was measured in EA.hy926 cells. (E) The stimulatory effect of Apo A-I on APC was confirmed by using primary human umbilical vein ECs. (F) Apo A-I (100-200 µg/mL) enhancement of APC barrier protection was titrated using the same assay. (G) To confirm that bioactive lipids potentially copurified with Apo A-I were not responsible for enhancement of APC barrier-protective function, the assay was repeated with either plasma-derived or recombinant Apo A-I (p-Apo A-I and r-Apo A-I, respectively; both 200 µg/mL). Experiments were performed in at least triplicate, and results are presented as the mean ± standard deviation. Statistical significance was determined by using the Student t test. *P < .05; **P < .01. n.s., not significant.
HDL and Apo A-I both enhance endothelial cell (EC) barrier protection by APC against thrombin-induced permeability. (A) The ability of the plasma lipoprotein HDL (1 mg/mL; red) to modulate protection against thrombin (5 nM) disruption of the EC barrier by APC (0.6-5 nM; blue) was measured. (B) Similarly, the ability of LDL (1 mg/mL) to mediate enhancement of APC barrier protection was tested under the same experimental conditions. (C) Evaluation of HDL and LDL enhancement of APC-mediated barrier protection was performed by titration of either HDL (blue) or LDL (red) in the presence of APC (5 nM) before thrombin (5 nM) exposure. (D) The ability of Apo A-I or Apo A-II (both 200 µg/mL) to enhance APC-mediated barrier protection was measured in EA.hy926 cells. (E) The stimulatory effect of Apo A-I on APC was confirmed by using primary human umbilical vein ECs. (F) Apo A-I (100-200 µg/mL) enhancement of APC barrier protection was titrated using the same assay. (G) To confirm that bioactive lipids potentially copurified with Apo A-I were not responsible for enhancement of APC barrier-protective function, the assay was repeated with either plasma-derived or recombinant Apo A-I (p-Apo A-I and r-Apo A-I, respectively; both 200 µg/mL). Experiments were performed in at least triplicate, and results are presented as the mean ± standard deviation. Statistical significance was determined by using the Student t test. *P < .05; **P < .01. n.s., not significant.
We next sought to investigate the molecular basis for HDL enhancement of APC cytoprotective activity, and therefore tested whether Apo A-I and Apo A-II, 2 abundant protein components found within HDL, might also mediate a similar effect to HDL when tested in purified form. Apo A-I but not Apo A-II was found to replicate the enhanced APC-dependent barrier function observed when APC was coincubated with HDL (Figure 1D). In addition, Apo A-I did not affect APC auto-degradation (supplemental Figure 1). To ensure that Apo A-I also mediated similar enhancement on primary endothelial cells, the same experiment was performed on human umbilical vein endothelial cells (Figure 1E). Similarly, no protection of endothelial barrier integrity from thrombin was observed in the presence of Apo A-I alone; however, Apo A-I significantly enhanced APC-mediated barrier protection, as before. Half-maximal barrier protection against thrombin-induced permeability was achieved by 50 to 100 µg/mL of Apo A-I, which is well within the normal physiological range for plasma Apo A-I (∼1.3 mg/mL) (Figure 1F). Copurified barrier-protective lipids were not responsible for the observed enhanced endothelial barrier protection,17,18 as both plasma-purified and recombinant Apo A-I exhibited identical enhancement of APC barrier protective function (Figure 1G).
Multiple studies have described the requirement for APC–EPCR binding to enable PAR1 signaling and protection of the endothelium from thrombin-induced barrier leakage.17,19,20 To assess how Apo A-I affects these requirements, we first performed the same assays in the presence of a PAR1 antagonist that blocks PAR1 signaling by APC (Figure 2A). The PAR1 antagonist blocked APC-mediated barrier protection irrespective of the presence of either HDL or Apo A-I. Similarly, an APC mutant with defective ability to mediate PAR1 proteolysis (APCE330A)20 remained ineffective in the presence of Apo A-I or HDL (Figure 2B). These data suggest that HDL or Apo A-I enhancement of APC cytoprotective activity on endothelial cells remains dependent on functional PAR1 signaling.

Apo A-I enhances endothelial cell (EC) barrier integrity and extracellular histone proteolysis by APC. (A) HDL- and Apo A-I–enhanced protection against thrombin-induced disruption of the EC barrier by APC (10 nM; blue) was measured in the presence of an anti-EPCR antibody (25 µg/mL, RCR-252) that blocks APC–EPCR binding (red) or with a PAR1 antagonist (SCH5300348; yellow) that prevents APC-dependent PAR1 signaling. (B) To confirm the role of PAR1 in Apo A-I–enhanced APC EC barrier protection, Apo A-I–dependent enhancement of either wild-type APC, or an APC variant that is unable to recognize PAR1 (APCE330A, both 10 nM), was characterized. (C) Similarly, the role of APC–EPCR binding in Apo A-I–stimulated APC signaling function was assessed by comparing protection of endothelial barrier integrity by wild-type APC and an APC variant unable to bind EPCR (APCL8F, both 10 nM). (D) Proteolysis of histone H3.1 by APC (6.25 nM) was assessed with HDL (125 µg/mL to 1 mg/mL) or Apo A-I (150 µg/mL) by western blotting with an antihistone H3.1 antibody, which recognizes both APC-cleaved and APC-uncleaved fragments of histone H3.1. (E) Similarly, proteolysis of histone H3.1 by titration of APC (3.25-25 nM) was assessed ± Apo A-I (150 µg/mL). (F) In addition, the ability of anionic phospholipids (20 µM) to modulate Apo A-I–mediated enhanced histone proteolysis by APC (6.25 nM) was assessed by using the same approach. Experiments were performed in at least triplicate, and results are presented as the mean ± standard deviation. Statistical significance was determined by using the Student t test. *P < .05. mAb, monoclonal antibody.
Apo A-I enhances endothelial cell (EC) barrier integrity and extracellular histone proteolysis by APC. (A) HDL- and Apo A-I–enhanced protection against thrombin-induced disruption of the EC barrier by APC (10 nM; blue) was measured in the presence of an anti-EPCR antibody (25 µg/mL, RCR-252) that blocks APC–EPCR binding (red) or with a PAR1 antagonist (SCH5300348; yellow) that prevents APC-dependent PAR1 signaling. (B) To confirm the role of PAR1 in Apo A-I–enhanced APC EC barrier protection, Apo A-I–dependent enhancement of either wild-type APC, or an APC variant that is unable to recognize PAR1 (APCE330A, both 10 nM), was characterized. (C) Similarly, the role of APC–EPCR binding in Apo A-I–stimulated APC signaling function was assessed by comparing protection of endothelial barrier integrity by wild-type APC and an APC variant unable to bind EPCR (APCL8F, both 10 nM). (D) Proteolysis of histone H3.1 by APC (6.25 nM) was assessed with HDL (125 µg/mL to 1 mg/mL) or Apo A-I (150 µg/mL) by western blotting with an antihistone H3.1 antibody, which recognizes both APC-cleaved and APC-uncleaved fragments of histone H3.1. (E) Similarly, proteolysis of histone H3.1 by titration of APC (3.25-25 nM) was assessed ± Apo A-I (150 µg/mL). (F) In addition, the ability of anionic phospholipids (20 µM) to modulate Apo A-I–mediated enhanced histone proteolysis by APC (6.25 nM) was assessed by using the same approach. Experiments were performed in at least triplicate, and results are presented as the mean ± standard deviation. Statistical significance was determined by using the Student t test. *P < .05. mAb, monoclonal antibody.
To investigate the requirement for APC–EPCR binding in Apo A-I–enhanced APC barrier protective function, barrier integrity was assessed in the presence of an anti-EPCR antibody (RCR-252) that blocks APC–EPCR binding and subsequent EPCR-dependent PAR1 proteolysis and signaling. As expected, the anti-EPCR antibody completely ablated the APC barrier-protective function. However, in the presence of Apo A-I or HDL, the requirement for EPCR binding was obviated, and APC barrier-protective function proceeded at an enhanced rate compared with wild-type APC (Figure 2A). To ensure that continued APC cytoprotective signaling did not occur due to residual APC–EPCR binding even in the presence of RCR-252, we compared the Apo A-I–enhanced barrier-protective function of wild-type APC with an APC mutant with defective EPCR-binding affinity (APCL8F). APCL8F was unable to mediate barrier protection from thrombin-induced barrier permeability. However, in the presence of either HDL or Apo A-I, APCL8F regained the ability to mediate protection against thrombin-induced barrier leakage and exhibited a similarly enhanced protective activity as wild-type APC (Figure 2C). Moreover, because sphingosine 1-phosphate receptor 1 (S1PR1) is essential for APC protection of endothelial barrier integrity,17,18 we evaluated APC protection from thrombin-induced barrier leakage in the presence of an S1PR1 inhibitor (Ex 26) but found that this impaired endothelial barrier protection by APC, both in the presence and absence of Apo A-I (supplemental Figure 2). These data indicate that the presence of Apo A-I promotes APC endothelial cell barrier protection from disruption using a mechanism that requires functional PAR1 and S1PR1 but not EPCR. This finding suggests that EPCR may not always be required for APC-induced endothelial cell barrier protection from thrombin-induced damage when Apo A-I/HDL is present.
Having shown that HDL and Apo A-I could increase PAR1-dependent endothelial barrier-protective activity, we next assessed whether HDL and Apo A-I could also modulate degradation of extracellular histones by APC. HDL was found to dose dependently enhance histone proteolysis by APC (Figure 2D). Incubation with Apo A-I alone had no effect (Figure 2E); however, coincubation of APC with Apo A-I caused significantly enhanced proteolysis of histone H3.1 by APC. Anionic phospholipid vesicles (PL) have previously been shown to increase histone proteolysis by APC9 but reportedly do not contribute to HDL enhancement of APC anticoagulant activity.14 Enhanced histone proteolysis by APC in the presence of Apo A-I was impaired by the presence of PL (Figure 2F), indicating Apo A-I and PL may compete for shared binding sites on APC.
Collectively, these studies describe a novel role for the Apo A-I component of HDL in enhancing APC inhibition of vascular dysfunction caused by endogenous barrier-disruptive and cytotoxic agents. The ability of Apo A-I/HDL to enhance multiple aspects of APC activities, including its anticoagulant, endothelial cell signaling and histone proteolytic activity, indicates that enhancement may be mediated by modulation of its proteolytic activity and suggests this activity may contribute to the beneficial therapeutic outcomes associated with APC administration.10-12 Moreover, the ability of HDL and Apo A-I to modulate APC endothelial cytoprotective activity suggests a potential role in modulating cardiovascular health outcomes in people with low HDL levels.
Data-sharing requests may be submitted to the corresponding author, Roger J. S. Preston (rogerpreston@rcsi.ie).
Acknowledgments
This study was supported by a Science Foundation Ireland Career Development Award (15/CDA/3499), a Technology and Innovation Development Award (18/TIDA/6001), and a National Children’s Research Centre Project Award (C/18/3).
Authorship
Contribution: All authors contributed to the experimental design, performed experiments and data analysis, and assisted with manuscript preparation and revision.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Roger J. S. Preston, Irish Centre for Vascular Biology, School of Pharmacy and Biomolecular Sciences, Royal College of Surgeons in Ireland, 123 St Stephen's Green, Dublin 2, Ireland; e-mail: rogerpreston@rcsi.ie.

