

Key Points
EC-targeted FVIII expression is more immunogenic than platelet-target expression.
ECs can present antigen FVIII to Tfh cells, promoting cell proliferation.

Abstract
Factor VIII (FVIII) replacement therapy for hemophilia A is complicated by development of inhibitory antibodies (inhibitors) in ∼30% of patients. Because endothelial cells (ECs) are the primary physiologic expression site, we probed the therapeutic potential of genetically restoring FVIII expression selectively in ECs in hemophilia A mice (FVIIInull). Expression of FVIII was driven by the Tie2 promoter in the context of lentivirus (LV)-mediated in situ transduction (T2F8LV) or embryonic stem cell–mediated transgenesis (T2F8Tg). Both endothelial expression approaches were associated with a strikingly robust immune response. Following in situ T2F8LV-mediated EC transduction, all FVIIInull mice developed inhibitors but had no detectable plasma FVIII. In the transgenic approach, the T2F8Tg mice had normalized plasma FVIII levels, but showed strong sensitivity to developing an FVIII immune response upon FVIII immunization. A single injection of FVIII with incomplete Freund adjuvant led to high titers of inhibitors and reduction of plasma FVIII to undetectable levels. Because ECs are putative major histocompatibility complex class II (MHCII)-expressing nonhematopoietic, “semiprofessional” antigen-presenting cells (APCs), we asked whether they might directly influence the FVIII immune responses. Imaging and flow cytometric studies confirmed that both murine and human ECs express MHCII and efficiently bind and take up FVIII protein in vitro. Moreover, microvascular ECs preconditioned ex vivo with inflammatory cytokines could functionally present exogenously taken-up FVIII to previously primed CD4+/CXCR5+ T follicular helper (Tfh) cells to drive FVIII-specific proliferation. Our results show an unanticipated immunogenicity of EC-expressed FVIII and suggest a context-dependent role for ECs in the regulation of inhibitors as auxiliary APCs for Tfh cells.
Introduction
Hemophilia A (HA) is a genetic bleeding disorder, resulting from a deficiency of factor VIII (FVIII). Although FVIII protein-replacement therapy is effective, 30% of severe HA patients will develop anti-FVIII inhibitory antibodies (inhibitors), rendering routine FVIII protein-replacement therapy useless. In such patients, immune-tolerance induction, infusing high quantities of FVIII, is the standard care. However, this approach costs over $1 million per patient-year and remains incompletely effective for 30% of inhibitor patients.1-5
Gene therapy is an attractive alternative for the treatment of HA. However, the potential to develop inhibitors to the neoprotein remains.B-15 Though not a natural site for FVIII expression, hepatocyte-targeted FVIII gene therapy has shown promising efficacy.16-18 Because platelets naturally expressed the FVIII carrier protein von Willebrand Factor (VWF), we previously engineered a lentiviral platelet-specific gene-therapy approach for on-demand delivery of FVIII at the site of injury.19 This partially restored hemostasis in HA mice and was associated with a degree of immunologic tolerance to infused FVIII protein.20-24 Recent studies confirm that the major physiologic site for FVIII synthesis is endothelial cells (ECs).25-30 Thus, we explored the potential for use of ECs as a target for gene therapy of HA.
Here, we investigated the efficacy of a lentivirus-based vector controlled by the EC-specific Tie2 promoter (T2F8) to restore FVIII expression in HA mice. We compared results of EC-targeted vs platelet-targeted FVIII expression using transgenic models. In both systems, we found endothelial-expressed FVIII to be surprisingly immunogenic. Our in vivo and ex vivo studies suggest that ECs can serve as auxiliary major histocompatibility complex class II (MHCII)–expressing antigen (Ag)-presenting cells (APCs) that mediate FVIII-specific stimulation of CD4+/CXCR5+ follicular T helper (Tfh) cells. These somewhat counterintuitive findings, together with previous studies, provide cautionary insight about endothelial-targeted gene therapy and support a putative site- and context-specific role for ECs as modulators of FVIII immunogenicity.
Materials and methods
Additional details on the reagents, methods, and statistics used in this study are provided in supplemental Material and methods.
Mice
Animal studies were approved by the institutional animal care and use committee of Medical College of Wisconsin or Harvard. Mouse models, as summarized in Table 1, included: (1) an FVIII-deficient (FVIIInull) model; (2) a platelet-specific FVIII expression model, 2bF8 transgenic (2bF8Tg) mice, in which human B-domain–deleted FVIII (hBDDFVIII) was expressed via the platelet-specific αIIb promotor31 ; (3) an EC-specific expression model, T2F8 transgenic (T2F8Tg) mice, in which hBDDFVIII was driven by the EC-specific Tie2 promoter32 ; (4) a CIITA−/− (MHCIInull) model; and (5) wild-type (WT) C57BL/6J mice.
Expression models/strains used in this study
| Model/strain name | Description | Deletions | Lentiviral expression | Transgenic expression | Genetic background |
|---|---|---|---|---|---|
| WT | Wild type | C57BL/6 | |||
| FVIIInull | Total mF8 deletion | mF8 | C57BL/6 | ||
| 2bF8LV | Total mF8 deletion, platelet-specific LV hF8 expression | mF8 | 2bF8LV via the αIIb promotor | B6/129S | |
| T2F8LV | Total mF8 deletion, endothelial-specific LV hF8 expression | mF8 | T2F8LV via the Tie-2 promotor | B6/129S | |
| 2bF8tg+/− | Total mF8 deletion, platelet-specific Tg hF8 expression | mF8 | hF8 via αIIb promotor | B6/129S | |
| T2F8tg+/− (line: ES#5) | Total mF8 deletion, EC-specific Tg hF8 expression (1 copy) | mF8 | hF8 via Tie-2 promotor | B6/129S | |
| T2F8tg+/+ (line: ES#5) | Total mF8 deletion, EC-specific Tg hF8 expression (2 copies) | mF8 | hF8 via Tie-2 promotor | B6/129S | |
| T2F8tg+/+ (line: ES #65) | Total mF8 deletion, EC-specific Tg hF8 expression (5 copies) | mF8 | hF8 via Tie-2 promotor | B6/129S | |
| T2F8TgF8−/−VWF−/− | Total mF8 and VWF deletions, EC-specific Tg hF8 expression | mF8, mVWF | hF8 via Tie-2 promotor | B6/129S | |
| CIITA−/− | Total CIITA deletion (MHCII null) | CIITA (MHCII transactivator) | C57BL/6 |
| Model/strain name | Description | Deletions | Lentiviral expression | Transgenic expression | Genetic background |
|---|---|---|---|---|---|
| WT | Wild type | C57BL/6 | |||
| FVIIInull | Total mF8 deletion | mF8 | C57BL/6 | ||
| 2bF8LV | Total mF8 deletion, platelet-specific LV hF8 expression | mF8 | 2bF8LV via the αIIb promotor | B6/129S | |
| T2F8LV | Total mF8 deletion, endothelial-specific LV hF8 expression | mF8 | T2F8LV via the Tie-2 promotor | B6/129S | |
| 2bF8tg+/− | Total mF8 deletion, platelet-specific Tg hF8 expression | mF8 | hF8 via αIIb promotor | B6/129S | |
| T2F8tg+/− (line: ES#5) | Total mF8 deletion, EC-specific Tg hF8 expression (1 copy) | mF8 | hF8 via Tie-2 promotor | B6/129S | |
| T2F8tg+/+ (line: ES#5) | Total mF8 deletion, EC-specific Tg hF8 expression (2 copies) | mF8 | hF8 via Tie-2 promotor | B6/129S | |
| T2F8tg+/+ (line: ES #65) | Total mF8 deletion, EC-specific Tg hF8 expression (5 copies) | mF8 | hF8 via Tie-2 promotor | B6/129S | |
| T2F8TgF8−/−VWF−/− | Total mF8 and VWF deletions, EC-specific Tg hF8 expression | mF8, mVWF | hF8 via Tie-2 promotor | B6/129S | |
| CIITA−/− | Total CIITA deletion (MHCII null) | CIITA (MHCII transactivator) | C57BL/6 |
.
In situ transduction of ECs in FVIIInull mice
We engineered a T2F8 lentiviral (LV) expression vector–harboring hBDDFVIII expression cassette under control of the Tie2 promoter (T2F8LV). T2F8LV was administered into FVIIInull mouse via tail-vein injection. Blood was collected for assessment of plasma FVIII levels and inhibitor titers.
Mouse immunization to human FVIII
FVIII immunization protocols were implemented as described.31-33 Briefly, for the T2F8Tg and 2bF8Tg groups, mice were immunized with recombinant hBDDFVIII (rhF8) at a dose of 600 U/kg plus incomplete Freund adjuvant (IFA) by intraperitoneal injection. WT mice were immunized with recombinant human full-length FVIII (rhfF8) at a dose of 600 U/kg with IFA via intraperitoneal injection. For FVIIInull mouse immunization, rhF8 was administered at a dose of 200 U/kg without IFA via IV injection.
Assays for measuring plasma CXCL13 in mice
WT C57BL/6 mice were immunized as in the previous section. Blood from immunized and unimmunized controls was collected 7 days after the last immunization and plasma CXCL13 was measured by enzyme-linked immunosorbent assay (ELISA).
Analysis of constitutive MHCII expression on ECs in vivo
Organs were harvested from untreated WT and CIITA−/− mice, minced, and collagenase digested. Cells were stained for CD31, CD105, CD11b, CD45, and MHCII and analyzed by flow cytometry as described.34,35 ECs were identified by gating on the CD31+/CD105+/CD11b−/CD45− cell population. Comparison of MHCII signal on CIITA−/− strain vs WT C57BL/6 was used to determine the specific MHCII signal.
In vitro study of CXCL13 expression and Tfh cell binding to ECs
Human dermal microvascular ECs (MVECs; DMVECs; hDMVECs) were cultured in the absence or presence of tumor necrosis factor α (TNF-α), interferon-γ (IFN-γ), interleukin 1β (IL1β), lipopolysaccharide (LPS), or recombinant human CXCL13 (rhCXCL13) and then washed and stained with anti-CXCL13 antibody. To study T-cell adhesion, LPS-pretreated hDMVECs were incubated with equal numbers of flow-sorted, human peripheral blood CD4+/CXCR5− or CD4+/CXCR5+ T cells for 1 hour in the absence and presence of function-blocking anti-CXCL13 antibody, followed by staining for CD4.
In vitro EC Ag-uptake and -processing studies
hDMVECs and murine DMVECs (mDMVECs) were treated with IFN-γ and TNF-α as described.36 To assess FVIII uptake, ECs incubated with rhfF8–Alexa 488 (10 U/mL) with human VWF (1 U/mL) and fibrinogen (FIB; 150 μg/mL) for 12 hours and with LysotrackerRed for 1 hour. To assess Ag processing by ECs, cells were incubated with the model Ag/APC-processing reporter DQ-OVA (full-length ovalbumin that exhibits proteolysis-dependent 488 nm37 ) and Lysotracker Red. Cells were washed and imaged live. As a more sensitive FVIII detection, rhfF8 was coated onto GMA-8021 (anti-FVIII monoclonal antibody [MAb])–conjugated carboxylate-modified fluorescent nanoparticles. Such “rhfF8 fluorospheres” were incubated with 150 μg/mL FIB (±1 U/mL VWF) and ECs for 1.5 hours followed by washing and imaging.
In vivo EC Ag-uptake and -processing studies
To investigate the ability of ECs to proteolytically process whole proteins in vivo, we immunized C57BL/6 mice subcutaneously in both flanks with 10 μg of DQ-OVA with complete Freund adjuvant. Four days later, ECs isolated from the skins around the immunization sites were analyzed for DQ-OVA.
ECs as APCs for T-cell proliferation
To test whether ECs can functionally present FVIII Ag, we performed a T-cell proliferation assay. Primary mDMVECs were cultured under conditions that recapitulate in vivo APC properties as established.36 Briefly, IFN-γ– and TNF-α–pretreated mDMVECs were cultured with CellTrace Violet–labeled CD4+ T cells isolated from rhF8-primed FVIIInull mouse spleens in media containing rhCXCL13 and rhF8 for 7 days. Cells were stained with anti-CD4, anti–T-cell receptor β (TCRβ), and anti-CXCR5 antibodies, and analyzed by flow cytometry.
Results
FVIIInull mice developed inhibitory antibodies after in situ lentivirus-mediated EC-specific transduction
Our previous studies showed that lentivirus-mediated platelet-specific gene delivery of FVIII (via αIIb promotor; 2bF8) can introduce sustained therapeutic levels of platelet FVIII expression and induce FVIII-specific immune tolerance in FVIIInull mice.20-22 However, levels of platelet FVIII in transduced recipients are low (∼5% to 10%) compared with healthy plasma.23 As an alternate approach, we performed lentivirus-mediated FVIII gene transfer with targeted expression in ECs via the panendothelial Tie2 promotor (T2F8LV). We confirmed that T2F8LV could introduce FVIII expression in ECs in vitro (supplemental Figure 1). Next, we introduced FVIII expression into ECs in vivo/in situ through systemic transduction of ECs in FVIIInull mice (Figure 1A). To our disappointment, no plasma FVIII was detected in T2F8LV-transduced animals (Figure 1B). However, all animals developed anti-FVIII inhibitors (Figure 1C), indicating that FVIII was expressed, but undetectable because of these antibodies. Total anti-FVIII immunoglobulin G (IgG) detected by ELISA was substantial and increased over 8 weeks (Figure 1D). These results suggest that in situ panendothelial transduction by T2F8LV with FVIII triggers strong anti-FVIII immune responses that sharply contrast the tolerance produced by lentivirus-mediated transduction of platelets,22-24 hepatocytes,16-18 and specific EC subsets.38,39
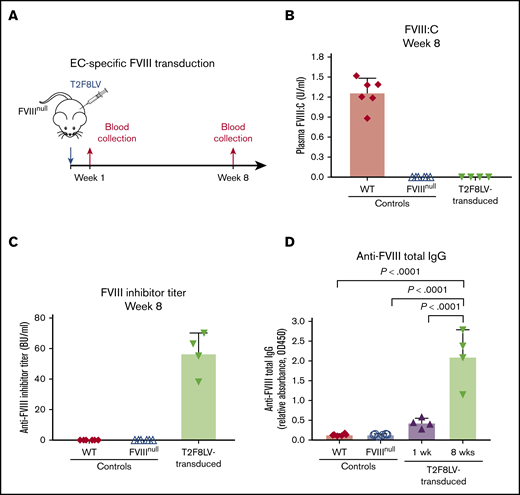
T2F8 LV-mediated in situ transduction of ECs in FVIIInullmice. (A) Schematic diagram of EC-specific FVIII transduction. Approximately 1 × 108 IU of T2F8LV in 300 µL of X-VIVO 10 media were administered into each 6-week-old FVIIInull mouse via tail-vein injection to transduce ECs in situ. Blood samples were collected following transduction, and plasmas were isolated for assays. Blood plasma from nontransduced WT C57BL/6 and naive FVIIInull mice served as controls. (B) Functional FVIII activity (FVIII:C) in plasma. Plasma FVIII:C levels in T2F8LV-transduced mice as determined by chromogenic assay. (C) Anti-FVIII inhibitor titers. Anti-FVIII inhibitor titers in WT, FVIIInull, and FVIIInull T2F8LV-transduced mice were determined by Bethesda assay. (D) Anti-FVIII total IgG in plasmas from T2F8LV-transduced mice as determined by ELISA. Data were analyzed using the 1-way analysis of variance followed by the Tukey test.
T2F8 LV-mediated in situ transduction of ECs in FVIIInullmice. (A) Schematic diagram of EC-specific FVIII transduction. Approximately 1 × 108 IU of T2F8LV in 300 µL of X-VIVO 10 media were administered into each 6-week-old FVIIInull mouse via tail-vein injection to transduce ECs in situ. Blood samples were collected following transduction, and plasmas were isolated for assays. Blood plasma from nontransduced WT C57BL/6 and naive FVIIInull mice served as controls. (B) Functional FVIII activity (FVIII:C) in plasma. Plasma FVIII:C levels in T2F8LV-transduced mice as determined by chromogenic assay. (C) Anti-FVIII inhibitor titers. Anti-FVIII inhibitor titers in WT, FVIIInull, and FVIIInull T2F8LV-transduced mice were determined by Bethesda assay. (D) Anti-FVIII total IgG in plasmas from T2F8LV-transduced mice as determined by ELISA. Data were analyzed using the 1-way analysis of variance followed by the Tukey test.
Immune responses are influenced by endothelial transgenic FVIII expression
To further evaluate the influence of EC-FVIII expression in immune responses, we prepared transgenic mice. Three groups of T2F8Tg mice from 2 lines generated by embryonic stem (ES) cell–mediated transgenesis32 were used for the FVIII immune response study (Table 1). One was the T2F8tg+/+ (ES#5) group, in which animals were homozygous T2F8Tg from line #5, with 100% normal levels of plasma FVIII (1.35 ± 0.12 U/mL). The second group of T2F8Tg mice was composed of heterozygous animals from line #5 (T2F8tg+/−; ES#5) with plasma FVIII coagulant activity (FVIII:C) of 0.58 ± 0.06 U/mL. In the third group of T2F8Tg mice were homozygous animals from another line (#65) (T2F8tg+/+; ES#65), with a FVIII:C level of 0.34 ± 0.14 U/mL.
We used an active immunization protocol31,32 to evaluate whether FVIII immunogenicity is diminished by endothelial expression. Surprisingly, we found that FVIII:C in the plasma dropped to undetectable levels in all 3 groups of T2F8Tg mice after a single immunization dose of rhF8 and IFA (Figure 2A-B). The plasma FVIII remained undetectable 4 weeks after this single immunization (not shown). In contrast, control WT C57BL/6 mice retained 69% of plasma FVIII after 1 immunization and still had detectable plasma FVIII even after a second immunization at week 4 (Figure 2A-B). These data suggest that targeted endothelial FVIII expression promotes strong immunogenicity, even if the expression is via germ line genetic modification.
![FVIII activity levels and anti-FVIII inhibitor titers in plasma of FVIII transgenic following FVIII immunization in the presence of IFA. (A) Immunization schemes for T2F8 and 2bF8 transgenic (left) and WT (control; right) mice. Preimmune blood was collected for all strains for FVIII:C assay. For T2F8 and 2bF8 transgenic mice that express hBDDFVIII, animals were immunized with rhF8 at 600 U/kg in the presence of IFA. Blood samples were collected 2 weeks after immunization. Because WT have full-length endogenous murine FVIII, mice received rhfF8 at 600 U/kg plus IFA and further blood collection was made the following week (week 3 overall). (B) Functional FVIII activity (FVIII:C) levels in T2F8 transgenic mice before and after immunization. FVIII:C levels were determined by chromogenic assay. Two lines of the endothelial-specific hBDDFVIII-expressing T2F8 transgenic mice with differing numbers of gene insertions (T2F8Tg[ES#5] and T2F8Tg[ES#65]) were used. WT mice were used as a control. (C) Comparison of inhibitor titers in T2F8 (EC-specific FVIII expression) and 2bF8 (platelet-specific FVIII expression) transgenic mice after a single dose of rhF8 immunization (week 2). The unpaired 2-tailed Student t test was used to analyze data.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/10/10.1182_bloodadvances.2020001468/2/m_advancesadv2020001468f2.png?Expires=1769184033&Signature=f6q-l9KXo28ZZh8xHz3MykweiIc8y8xI9XzIyZ~onw22MwyLhGxLmz7OGew40afMQrePeBAmGD7-QTiNr2nKL5fqH5Ruo4R4z3WIf8Mk5LZbx-ryUO~kwg5pCxPF4y2AqTqOu8mAkLlKwskTZjPUn-ZVXTM37KN0fq16Yu0vXd2Qz4l0KKS6xqAjKbZX5xMOmGxj5PkTYnGd-r4yS6OuU591akvxWHST2Ff13tVrPo0E6DhIgobiEkVO6ur6sNSKQBWYsQiSzibVaBj4jWZVf24oWNgFrn46cpnM44X5Zxs7gNPOpr7hs6yi~VgpclgFXkqJLgrKR2BAAb5fPbJt1w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
FVIII activity levels and anti-FVIII inhibitor titers in plasma of FVIII transgenic following FVIII immunization in the presence of IFA. (A) Immunization schemes for T2F8 and 2bF8 transgenic (left) and WT (control; right) mice. Preimmune blood was collected for all strains for FVIII:C assay. For T2F8 and 2bF8 transgenic mice that express hBDDFVIII, animals were immunized with rhF8 at 600 U/kg in the presence of IFA. Blood samples were collected 2 weeks after immunization. Because WT have full-length endogenous murine FVIII, mice received rhfF8 at 600 U/kg plus IFA and further blood collection was made the following week (week 3 overall). (B) Functional FVIII activity (FVIII:C) levels in T2F8 transgenic mice before and after immunization. FVIII:C levels were determined by chromogenic assay. Two lines of the endothelial-specific hBDDFVIII-expressing T2F8 transgenic mice with differing numbers of gene insertions (T2F8Tg[ES#5] and T2F8Tg[ES#65]) were used. WT mice were used as a control. (C) Comparison of inhibitor titers in T2F8 (EC-specific FVIII expression) and 2bF8 (platelet-specific FVIII expression) transgenic mice after a single dose of rhF8 immunization (week 2). The unpaired 2-tailed Student t test was used to analyze data.
FVIII activity levels and anti-FVIII inhibitor titers in plasma of FVIII transgenic following FVIII immunization in the presence of IFA. (A) Immunization schemes for T2F8 and 2bF8 transgenic (left) and WT (control; right) mice. Preimmune blood was collected for all strains for FVIII:C assay. For T2F8 and 2bF8 transgenic mice that express hBDDFVIII, animals were immunized with rhF8 at 600 U/kg in the presence of IFA. Blood samples were collected 2 weeks after immunization. Because WT have full-length endogenous murine FVIII, mice received rhfF8 at 600 U/kg plus IFA and further blood collection was made the following week (week 3 overall). (B) Functional FVIII activity (FVIII:C) levels in T2F8 transgenic mice before and after immunization. FVIII:C levels were determined by chromogenic assay. Two lines of the endothelial-specific hBDDFVIII-expressing T2F8 transgenic mice with differing numbers of gene insertions (T2F8Tg[ES#5] and T2F8Tg[ES#65]) were used. WT mice were used as a control. (C) Comparison of inhibitor titers in T2F8 (EC-specific FVIII expression) and 2bF8 (platelet-specific FVIII expression) transgenic mice after a single dose of rhF8 immunization (week 2). The unpaired 2-tailed Student t test was used to analyze data.
After a single rhF8/IFA immunization, there were no statistically significant differences in inhibitor titers between the different groups of T2F8 mice in the FVIIInull background (supplemental Figure 2A). This indicates that FVIII-inhibitor formation in mice with endothelial expression is not substantially influenced by the level of FVIII expression over this range. We asked whether VWF might influence inhibitor development (supplemental Figure 2B). The inhibitor titer in the T2F8tg+/+VWF−/− group (252 ± 144 BU/mL) was significantly lower than the T2F8tg+/+ group (810 ± 508 BU/mL; supplemental Figure 2B), although plasma FVIII:C in the T2F8tg+/+VWF−/− group was not detectable (not shown). After the second FVIII immunization, the inhibitor titers in the T2F8tg+/+ group (8560 ± 7074 BU/mL) were significantly higher than in the WT control group (2576 ± 4224 BU/mL) (supplemental Figure 2C).
We then compared FVIII immune responses in the endothelial-expressed, T2F8Tg, and platelet-expressed 2bF8Tg models (Figure 2C). The 2bF8Tg mice used in this study were also generated by ES cell–mediated transgenesis.31 In 2bF8Tg mice, FVIII expression is targeted to platelet α-granules with a level of ∼0.75 mU/108 platelets, which corresponds to 1.2% of FVIII in WT mouse whole blood (though no plasma FVIII:C was detectable31 ). When the same immunization protocol was applied to our platelet-specific FVIII (2bF8Tg) transgenic mice, 15 of 18 2bF8Tg+/− mice developed inhibitors with a titer of 54 ± 49 BU/mL. All T2F8Tg mice developed inhibitors with the average titer of 1811 ± 1953 BU/mL, which was significantly higher than obtained in 2bF8Tg mice (Figure 2C). Thus, expression in ECs greatly enhances immunogenicity of FVIII, whereas expression in megakaryocytes/platelets induces a degree of immune tolerance.
ECs exhibit functional properties of APCs
B-cell and antibody responses are tightly regulated by the CD4+/CXCR5+ lymph node Tfh cell subset, which in turn is regulated by MHCII-expressing APCs.40-46 Because ECs have emerged as auxiliary MHCII-expressing APCs,47,48 we hypothesized that they might directly influence FVIII immunogenicity by presenting MHCII/Ag to Tfh cells. Human ECs are well established to constitutively express MHCII in a response to tonic levels of IFN-γ.47,49 In mice the situation remains less studied and less clear.50,51 Therefore, we first examined whether murine ECs express MHCII at steady state. We conducted flow cytometric analysis of ECs freshly isolated from a range of tissues in healthy C57BL/6 WT or CIITA−/− (MHCIInull) mice. EC-specific MHCII was measured by gating on the CD31+/CD105+/CD11b−/CD45− cell population (supplemental Figure 3A). As shown in supplemental Figure 3B, ECs of heart, lung, kidney, liver, and skin all showed strong constitutive levels of MHCII in WT mice as compared with CIITA−/− mice. These results suggest that, like human ECs, murine ECs exhibit broad constitutive IFN-γ–/CIITA-dependent MHCII expression and have potential to function as auxiliary class II APCs in vivo. Of note, all cultured ECs require addition of exogenous IFN-γ to support in vitro MHCII expression36,47,51 (as we confirm in supplemental Figure 4A-B).
CXCL13 supports interaction of ECs with circulating Tfh cells
A defining feature of Tfh is expression of the CXCL13-selective chemokine receptor CXCR5, which orchestrates its functions within lymphoid organs.40-46 Circulating CD4+/CXCR5+ Tfh cells with memory-like properties (cTfh cells), as well as CXCL13, are also found in the blood with elevated levels seen during humoral and inflammatory responses.52-60 We hypothesized that cTfh cells might interact with vascular ECs in a CXCL13-promoted manner. We first examined blood plasma levels of CXCL13 in mice following rhfF8/IFA immunization during inhibitor formation, and found a substantial, approximately fivefold, increase in the immunized vs control mice. (Figure 3A). Because ECs can bind chemokines via cell-surface heparan sulfates,61,62 we tested whether exogenous CXCL13 could be “captured” by ECs. Brief incubation of resting ECs with rhCXCL13 followed by staining and imaging revealed robust levels of cell-surface–captured CXCL13 (Figure 3B-C). ECs also have potential to express CXCL13 under inflammatory conditions,63-67 and LPS is known to promote CXCL13 expression by professional APCs.68,69 Thus, we treated hDMVECs with LPS or several inflammatory cytokines (ie, TNF-α, IFN-γ, IL1β). We found that the former induced substantial expression and surface presentation of endogenous CXCL13 (Figure 3B-C). In parallel studies, we also confirmed previous studies indicating that LPS, TNF-α, and IL1β (but not IFN-γ) upregulated ICAM-1, a representative proinflammatory immune-adhesion molecule (supplemental Figure 4C-D).
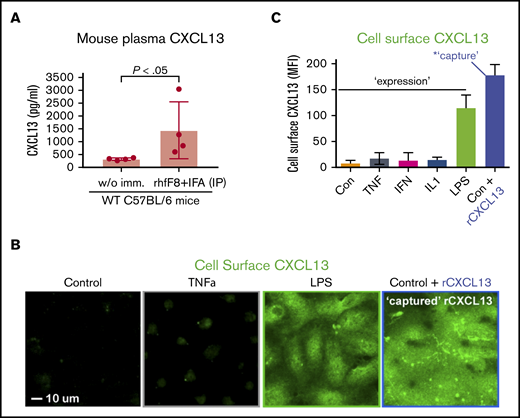
Circulating CXCL13 in vivo and CXCL13 expression and presentation by ECs in vitro. (A) WT C57BL/6 mice were immunized with rhfF8 by intraperitoneal injection with IFA. Blood samples from immunized and paired unimmunized controls were collected 7 days later and plasma CXCL13 was measured by ELISA. (B-C) hDMVECs were cultures in the absence (control) or presence of 100 ng/mL recombinant human inflammatory cytokines (TNF-α, IFN-γ, IL1-β) or 10 ng/mL LPS for 24 hours. Alternatively, untreated control hDMVECs were incubated with 100 ng/mL recombinant human CXCL13 for 1 hour. Cells were then rinsed, fixed, and stained (without permeabilization) with anti-CXCL13 antibody. (B) Representative images (for select conditions) with anti-CXCL13 antibody staining shown in green. Scale bar, 10 μm. (C) For each condition, mean fluorescence intensity (MFI) of CXCL13 staining (representing endogenous expression and cell-surface presentation of CXCL13 or capture/presentation of exogenous CXCL13) was quantified per field of view for a total of 10 fields of view for each of 2 separate experiments and averaged.
Circulating CXCL13 in vivo and CXCL13 expression and presentation by ECs in vitro. (A) WT C57BL/6 mice were immunized with rhfF8 by intraperitoneal injection with IFA. Blood samples from immunized and paired unimmunized controls were collected 7 days later and plasma CXCL13 was measured by ELISA. (B-C) hDMVECs were cultures in the absence (control) or presence of 100 ng/mL recombinant human inflammatory cytokines (TNF-α, IFN-γ, IL1-β) or 10 ng/mL LPS for 24 hours. Alternatively, untreated control hDMVECs were incubated with 100 ng/mL recombinant human CXCL13 for 1 hour. Cells were then rinsed, fixed, and stained (without permeabilization) with anti-CXCL13 antibody. (B) Representative images (for select conditions) with anti-CXCL13 antibody staining shown in green. Scale bar, 10 μm. (C) For each condition, mean fluorescence intensity (MFI) of CXCL13 staining (representing endogenous expression and cell-surface presentation of CXCL13 or capture/presentation of exogenous CXCL13) was quantified per field of view for a total of 10 fields of view for each of 2 separate experiments and averaged.
To assess the function of EC-presented CXCL13 on cTfh cell adhesion, we incubated LPS-pretreated hDMVECs with sorted human peripheral blood mononuclear cell (PBMC)–derived CD4+/CXCR5− (conventional) and CD4+/CXCR5+ (cTfh) T cells in the absence and presence of a CXCL13 function-blocking antibody (Figure 4A). Under these conditions, the level of CD4+/CXCR5+ cTfh binding to ECs was approximately threefold greater than that of the conventional CD4+/CXCR5− T cells (Figure 4B). Anti-CXCL13 antibody largely blocked binding of CXCR5+CD4+, but not the CD4+/CXCR5−, T cells. These data indicate that binding of CD4+/CXCR5+ cTfh cells to ECs can be promoted by CXCL13.
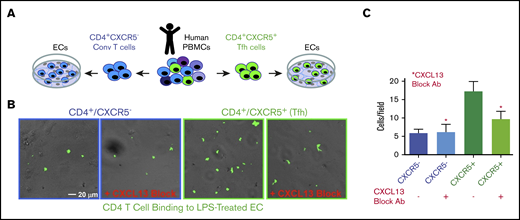
Adhesion of conventional and CXCR5+CD4+T cells to LPS-activated hDMVECs. (A) Human peripheral blood CD4+/CXCR5− (conventional) or CD4+/CXCR5+ (cTf) T cells flow sorted. Equal numbers of CD4+/CXCR5− and CD4+/CXCR5+ T cells were then incubated for 1 hour on hDMVECs that were pretreated for 24 hours with LPS (10 ng/mL) in the absence and presence of 20 μg/mL function-blocking anti-CXCL13 antibody. Samples were then subjected to brief washing with warm media, fixation, staining for CD4, and imaging. (B) Representative fields of view showing phase and fluorescence images overlaid. T cells were labeled with anti-CD4-Alexa488 antibody, shown in green. Scale bar, 20 μm. (C) Quantitation of average number of cells per field of view (10 images per condition for 2 separate experiments).
Adhesion of conventional and CXCR5+CD4+T cells to LPS-activated hDMVECs. (A) Human peripheral blood CD4+/CXCR5− (conventional) or CD4+/CXCR5+ (cTf) T cells flow sorted. Equal numbers of CD4+/CXCR5− and CD4+/CXCR5+ T cells were then incubated for 1 hour on hDMVECs that were pretreated for 24 hours with LPS (10 ng/mL) in the absence and presence of 20 μg/mL function-blocking anti-CXCL13 antibody. Samples were then subjected to brief washing with warm media, fixation, staining for CD4, and imaging. (B) Representative fields of view showing phase and fluorescence images overlaid. T cells were labeled with anti-CD4-Alexa488 antibody, shown in green. Scale bar, 20 μm. (C) Quantitation of average number of cells per field of view (10 images per condition for 2 separate experiments).
ECs take up and process protein Ag, including FVIII, in vitro and in vivo
ECs might process endogenously expressed FVIII that is diverted from the secretory pathway as well as exogenous FVIII that is actively taken up by endocytosis.70-77 To evaluate the latter, we incubated rhfF8–Alexa 488 with conditioned human DMVECs in the presence of VWF and FIB. rhfF8–Alexa 488 was readily taken up into perinuclear clusters that colocalized with late endosomes and lysosomes, features consistent with class II processing (Figure 5A). Similar observations were made with conditioned mDMVECs (not shown). Complementary experiments were performed using rhfF8-labeled with 40-nm fluorospheres that provide an enhanced signal-to-noise ratio. Upon incubation with conditioned mDMVECs in the presence or absence of VWF, these “rhfF8 fluorospheres” bound to membrane surface patches and accumulated in putative intracellular perinuclear puncta (presumptive endosomes/lysosomes; Figure 5B left panel). VWF did not alter the pattern of binding or uptake (not shown). Little binding or uptake was visible on ECs incubated with control, non–rhfF8-coupled fluorospheres (Figure 5B right panel). Both methods indicated that FVIII is taken up by a majority of ECs into a perinuclear compartment.
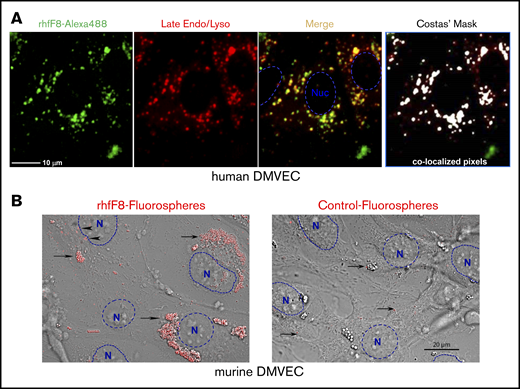
Binding and uptake of FVIII by conditioned DMVECs. (A) Confluent hDMVECs were pretreated with 100 nM IFN-γ (48 hours) and 100 nM TNF-α (24 hours) prior to incubation for 12 hours rhfF8–Alexa 488 (10 U/mL; in the presence of 1 U/mL VWF and 150 µg/mL FIB) together with Lysotracker Red (late endosome/lysosome marker; 50 nM). Cells were then rinsed and imaged live. Fluorescence microscopy showed significant rhfF8–Alexa 488 (green) accumulated into perinuclear punctae, which partially colocalized with late endosomes/lysosomes (red) as seen by the yellow signal in the merged image. This was validated by computational image analysis, which also identified substantial areas of rhfF8-488 and Lysotracker Red coincidence, as depicted by white pixels in the Costes mask (right panel) and reflected in an average Pearson correlation coefficient of 0.680 ± 0.033 (r2 > 0.98; Costes P = 100%). Scale bar, 25 μm. (B) As a complementary and more sensitive approach to detect FVIII binding and uptake, rhfF8 was coated onto GMA-8021 (anti-FVIII MAb)-conjugated carboxylate-modified red fluorescent nanoparticles (rhfF8 fluorospheres). The same fluorescent particles were subjected to blocking of the carboxylate modification with 2% BSA to demonstrate the level of nonspecific binding and cellular uptake (Control fluorospheres). Fluorospheres were then incubated together with 150 µg/mL fibrinogen in the presence (not shown) or absence of 1 U/mL VWF for 1.5 hours on IFN-γ– and TNF-α–conditioned mDMVECs. Differential interference contrast (DIC) and confocal fluorescence images were superimposed. Arrowheads indicate patches of putative membrane-bound rhfF8. Arrows indicate putatively internalized perinuclear clusters of rhfF8 that presumably represent endosomes and lysosomes (left panel). The same pattern was observed in the presence of VWF. In the absence of rhfF8 coating, control fluorospheres showed very little EC binding or uptake (right panel, arrows). Scale bar, 20 μm.
Binding and uptake of FVIII by conditioned DMVECs. (A) Confluent hDMVECs were pretreated with 100 nM IFN-γ (48 hours) and 100 nM TNF-α (24 hours) prior to incubation for 12 hours rhfF8–Alexa 488 (10 U/mL; in the presence of 1 U/mL VWF and 150 µg/mL FIB) together with Lysotracker Red (late endosome/lysosome marker; 50 nM). Cells were then rinsed and imaged live. Fluorescence microscopy showed significant rhfF8–Alexa 488 (green) accumulated into perinuclear punctae, which partially colocalized with late endosomes/lysosomes (red) as seen by the yellow signal in the merged image. This was validated by computational image analysis, which also identified substantial areas of rhfF8-488 and Lysotracker Red coincidence, as depicted by white pixels in the Costes mask (right panel) and reflected in an average Pearson correlation coefficient of 0.680 ± 0.033 (r2 > 0.98; Costes P = 100%). Scale bar, 25 μm. (B) As a complementary and more sensitive approach to detect FVIII binding and uptake, rhfF8 was coated onto GMA-8021 (anti-FVIII MAb)-conjugated carboxylate-modified red fluorescent nanoparticles (rhfF8 fluorospheres). The same fluorescent particles were subjected to blocking of the carboxylate modification with 2% BSA to demonstrate the level of nonspecific binding and cellular uptake (Control fluorospheres). Fluorospheres were then incubated together with 150 µg/mL fibrinogen in the presence (not shown) or absence of 1 U/mL VWF for 1.5 hours on IFN-γ– and TNF-α–conditioned mDMVECs. Differential interference contrast (DIC) and confocal fluorescence images were superimposed. Arrowheads indicate patches of putative membrane-bound rhfF8. Arrows indicate putatively internalized perinuclear clusters of rhfF8 that presumably represent endosomes and lysosomes (left panel). The same pattern was observed in the presence of VWF. In the absence of rhfF8 coating, control fluorospheres showed very little EC binding or uptake (right panel, arrows). Scale bar, 20 μm.
Several studies support the ability of ECs to mediate functional class II Ag processing.70-74 To extend this idea, we implement DQ-OVA, a common Ag-processing reporter.37,78 We incubated hDMVECs (supplemental Figure 5A) and mouse heart MVECs (not shown) with DQ-OVA together with a LysoTracker Red. DQ-OVA was readily taken up into ECs and converted to fluorescent peptides that partially colocalized with late endosomes/lysosomes (supplemental Figure 5A). We extended these studies into the in vivo setting by subcutaneous immunization of DQ-OVA in C57BL/6. Injection-site skin was later harvested and analyzed by flow cytometry. This showed substantial levels of processed DQ-OVA signal in ECs (supplemental Figure 5B). Together, these studies show that whole protein Ag is readily taken up and proteolytically processed by human and murine ECs both in vitro and in vivo.
ECs can mediate functional FVIII-dependent stimulation of Tfh
To determine whether murine ECs can promote Ag-specific immune responses in CD4+/CXRC5+ Tfh cells, we performed a T-cell proliferation assay (Figure 6B). CD4+ T cells isolated from FVIII-primed mice were cocultured ex vivo with ECs and CXCL13, with or without rhF8. Proliferating T-cell clusters/clones were seen only in cocultures that contained rhF8 Ag (not shown). Flow cytometry showed that ∼7% of the FVIII-primed CD4+ T cells were proliferating (daughter) when restimulated with ECs with either 0.2 μg/mL or 2 μg/mL rhF8 (Figure 6B). In contrast, only 1% of T cells proliferated when cocultured in the absence of rhF8 or cocultured with an unrelated Ag rhF9 (2 μg/mL; Figure 6B). When the CD4+/CD5+ Tfh cells were selectively analyzed, there were 18% and 12.7% Tfh proliferating daughter cells in cocultures containing 0.2 and 2 μg/mL rhF8, respectively. In contrast, there were only 5% Tfh daughter cells when cocultured without rhF8 or with 2 μg/mL rhF9 control Ag (Figure 6B). To confirm that the isolated FVIII-primed CD4+ T-cell population did not contain APCs, we cultured isolated CD4+ T cells with rhF8 in the absence of ECs and saw no detectable proliferation in either the total CD4+ T or Tfh cell populations (supplemental Figure 6). These data confirm that ECs can take up FVIII and functionally present FVIII Ag to FVIII-primed CD4+ T cells, leading to Ag-selective Tfh cell proliferation.
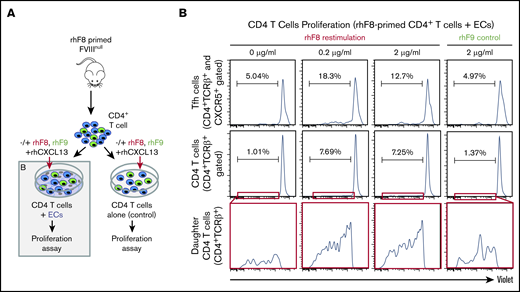
Ag-specific CD4 T-cell proliferation assay. (A) Schematic diagram of T-cell proliferation assay. CD4+ T cells isolated from rhF8-primed FVIIInull mouse were labeled with CellTrace Violet and cocultured with DMVECs that were pretreated with IFN-γ/TNF-α in the presence of CXCL13 and the absence and presence of rhF8 at 37°C in 5% CO2 for 1 week. rhF9 was used as an unrelated Ag control. CD4 T cells plated in the absence of endothelium served as an additional control (supplemental Figure 6). (B) Flow cytometry analysis daughter (proliferated) cells. Cells were stained for CD4, TCRβ, and CXCR5, and analyzed by flow cytometry. Dead cells were excluded by the 7-AAD staining. CD4 T-cell divisions were quantified as the dilution of CellTrace Violet signal in daughter cells away from the initially labeled peak. Results for both CD4 Tfh (top row) and total CD4 (middle row) are shown. Bottom row shows expanded view of daughter cell in the red, boxed regions from the middle row. These data demonstrate that ECs can present FVIII to Tfh cells, promoting Ag-specific cell proliferation.
Ag-specific CD4 T-cell proliferation assay. (A) Schematic diagram of T-cell proliferation assay. CD4+ T cells isolated from rhF8-primed FVIIInull mouse were labeled with CellTrace Violet and cocultured with DMVECs that were pretreated with IFN-γ/TNF-α in the presence of CXCL13 and the absence and presence of rhF8 at 37°C in 5% CO2 for 1 week. rhF9 was used as an unrelated Ag control. CD4 T cells plated in the absence of endothelium served as an additional control (supplemental Figure 6). (B) Flow cytometry analysis daughter (proliferated) cells. Cells were stained for CD4, TCRβ, and CXCR5, and analyzed by flow cytometry. Dead cells were excluded by the 7-AAD staining. CD4 T-cell divisions were quantified as the dilution of CellTrace Violet signal in daughter cells away from the initially labeled peak. Results for both CD4 Tfh (top row) and total CD4 (middle row) are shown. Bottom row shows expanded view of daughter cell in the red, boxed regions from the middle row. These data demonstrate that ECs can present FVIII to Tfh cells, promoting Ag-specific cell proliferation.
Discussion
Our experiments demonstrate that ECs have an unexpected capacity to enhance anti-FVIII antibody formation in HA mice. This was true whether FVIII gene expression was via LV-mediated gene therapy or by germ line expression in transgenic mice. Our supporting ex vivo/in vitro studies led us to hypothesize a direct pathway through which inflammatory-conditioned ECs can act as auxiliary APCs in FVIII immune responses. These findings have relevance for gene therapy and possibly for understanding more generally the unusual immunogenicity of FVIII.
ECs are known as the major physiologic site of FVIII expression25-30 and anti-FVIII antibodies are rare in healthy subjects.79 As such, our observation of strong anti-FVIII immunological responses to EC-targeted therapeutic expression of FVIII seems rather counterintuitive. We view this as an apparent, rather than an actual, conflict. Our data bear directly on engineered FVIII expression, whereby synthesis was driven in all ECs via the panendothelial Tie2 promotor. Native FVIII production was recently shown to be restricted to a smaller subset of ECs that primarily include liver sinusoidal (LSEC), lymphatic (LEC), and possibly splenic sinusoidal (SSEC) ECs.25,26,29,30 It follows that the ectopic expression of FVIII within nonnative ECs (ie, ECs other than LSEC, LEC, and SSEC) is responsible for the observed immunogenicity, and this seems to be specific for ECs, as ectopic nonnative expression of FVIII in hepatocytes16-18 and platelets20-24,80 are both tolerated.
Previous studies are consistent with this dichotomy among EC types. We reported that transgenic panendothelial expression of hFVIII (eg, confirmed in lung and heart ECs) in HA mice had heightened responses to hFVIII immunization compared with mice with platelet expression of FVIII.32 Similarly, use of the “sleeping beauty transposon” to express FVIII specifically in lung ECs was immunogenic,81 whereas use of the same FVIII transposon system targeted to LSEC promoted tolerance.82 Finally, 2 recent reports described transduction strategies that were similar to our T2F8LV, but used either the native FVIII38 or the “vascular endothelial cadherin”39 promoter. FVIII expression in both cases was restricted largely to liver ECs (ie, LSEC) and was not found in ECs of the lung or kidney.38,39 In these studies, FVIII was restored to ∼25% and ∼5%, respectively, of normal and was associated with degree of immune tolerance.38,39 Taken together, these reports suggest that ectopic expression of FVIII in hepatocytes and platelets seems relatively nonimmunogenic and partially tolerogenic. In contrast, endothelial expression of FVIII leads to either enhanced immunogenicity or tolerance, depending on the specific endothelial location or subsets that are targeted.
We hypothesized that the mechanism lies in the ability of ECs to function directly as APCs with site- and context-specific properties.47,48 Contrasting with hepatocytes and platelets, all ECs in both humans and mice (eg, shown herein and in Choo et al50 and in Kreisel et al51 ) constitutively express MHCII and can mediate class II presentation of both endogenous and exogenous Ags.47-49,73 LSEC, LEC, where most FVIII is normally expressed, exhibit unique constitutive protolerogenic properties (eg, expression of programmed death ligand 1 [PD-L1], transforming growth factor [TGF-β]), which would be expected to promote Ag-specific tolerance to FVIII.83-89 Alternatively, the other nonnative ECs tend to stimulate Ag-specific responses, particularly in inflammatory settings.48,72,90-93 Our findings with engineered panendothelial expression of FVIII in both native and nonnative ECs suggest that the proimmunogenic potential of the latter can exert a dominant stimulatory regulation on FVIII inhibitors (ie, broken immunological tolerance).
Development of FVIII inhibitors requires professional APCs, such as lymphoid dendritic cells, to prime naive CD4+ T cells and initiate formation of Ag-specific memory CD4+/CXCR5+ Tfh cells.94-104 Our data do not conflict with this model. Rather, they indicate that ECs can act as complementary APCs that modulate response of memory-like cTfh cells after they have been primed by lymphoid dendritic cells entering the blood. Studies in other contexts suggest that cTfh cells receive distinct regulatory cues within the circulation.53,55-57,105-107 cTfh cells ultimately traffic back to lymph nodes and spleen, with robust cytokine responsiveness and presumably altered capacity for directing antibody formation.53,55-57 Thus, we propose that inflammatory-conditioned, particularly nonnative, ECs displaying FVIII Ag can promote the maturation and function of FVIII-specific cTfh cells.
Our experiments support this hypothesis by showing that panendothelial, but not platelet, expression upregulates the immunogenicity of FVIII in mice under inflammatory settings. We show that ECs express and bind CXCL13 in response to LPS and that this promotes adhesion of Tfh cells. We show that inflammatory-conditioned ECs take up and traffic FVIII consistently with class II Ag processing. Finally, we show that CD4+/CXCR5+ Tfh cells from FVIII-immunized mice proliferate in response to ex vivo presentation of FVIII by ECs. These observations support the plausibility of our hypothesis, though more work is needed to establish the extent to which this pathway may influence development of anti-FVIII antibodies.
In mice, immunogenicity was highest for the T2F8LV-mediated in situ EC transduction. We speculate that ECs sense proinflammatory danger signals108 and/or endoplasmic reticulum stress generated by the viral vector delivering of T2F8, which upregulates their immunogenic APC functions and promotes FVIII humoral response. Inhibitors were detected in T2F8LV-transduced FVIIInull mice even when plasma FVIII:C was undetectable. Immune responses in the transgenic and WT controls required an active immunization with FVIII together with IFA. Response of WT control mice to rhfF8/IFA is likely to be explained by species differences in FVIII epitopes. However, this does not explain the response of T2F8Tg mice, which are born with normal levels of rhF8 and should be tolerized to rhF8. In vivo Ag presentation by ECs has been tested using a Tie2-driven EC-specific β-gal transgenic mouse model.73 Studies with this model suggest that ECs can present intracellular self-Ag to the immune system without completely deleting Ag-specific T cells.73 Apparently, EC-expressed FVIII results in immunological ignorance rather than tolerance, implying that EC presentation during T-cell development allows escape of FVIII-specific CD4+ T cells in T2F8Tg mice.
Our data suggest the potential immunogenic roles of FVIII Ag presentation by ECs in the context of engineered gene therapy. It is attractive to speculate that ECs could also modulate FVIII immune responses to exogenous FVIII (eg, HA therapy). LSEC, LEC, and SSEC constitutively express FVIII/VWF-scavenging receptors/activity. Under inflammatory conditions, many other ECs can upregulate expression of MHCII and costimulatory molecules, as well as scavenging properties such as stabilin-2.75-77,109-113 Moreover, in contrast to dendritic cells, ECs are exposed to circulating VWF and are positioned to distinguish between multimer size and conformation. Because these factors appear to influence immunogenicity of FVIII, it is tempting to speculate that endothelial Ag presentation plays a broader role in FVIII immunogenicity.114,115 Further investigation of the role of EC subsets in modulating FVIII immune responses is warranted.
We report binding, uptake, and presentation of FVIII by inflammatory conditioned DMVECs. Although binding of FVIII to ECs has previously been reported, the mechanism(s) remain poorly characterized. FVIII may bind directly to ECs, presumably binding through exposed phosphatidylserine,116 enabling assembly of the intrinsic FXa complex. FVIII may bind indirectly through VWF and possibly fibrin(ogen)117 via stabilin-2.118 VWF also binds ECs via P-selectin and αvβ3 integrin,119,120 as well as other scavenger receptors aimed at molecular clearance and recycling.118 FVIII has been shown to bind to lipoprotein-related protein and other scavenger receptors shared by ECs and other cell types.121 In addition, there are other candidate FVIII-binding molecules, particularly SREC-1, a scavenger receptor that is partially restricted to ECs.122 Further studies will be needed to identify the receptor and the uptake pathway responsible for FVIII uptake in the MVECs.
In summary, our data demonstrate that panendothelial targeting of FVIII is more immunogenic than expression by megakaryocytes (or hepatocytes), implying a novel role of ECs in modulation of the humoral immune response to FVIII. We showed for the first time the FVIII uptake by MVECs and functional FVIII presentation by MVECs to FVIII-primed Tfh cells, leading to cell proliferation. Together, our results indicate that ECs may play an important role in FVIII immune responses.
Data-sharing requests may be e-mailed to the corresponding author, Qizhen Shi (qshi@versiti.org).
Acknowledgments
The authors thank the Harvard Center for Biological Imaging for infrastructure and support, Haig H. Kazazian at the University of Pennsylvania School of Medicine for the FVIIInull mice, and Linzheng Shi at the Boston University School of Medicine for help designing some of the schematic diagrams in their studies. The VWF−/− mice were developed by Denisa D. Wagner at Harvard Medical School and purchased from The Jackson Laboratory.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants R01HL102035 (Q.S.), R01HL104006 (C.V.C.), and R56HL111640 (C.V.C.), a National Hemophilia Foundation Bridge Award (Q.S.), and generous gifts from the Children’s Hospital of Wisconsin Foundation (Q.S.), the Midwest Athletes Against Childhood Cancer Fund (Q.S.), and by a Merit Award of the Veterans Administration (G.E.G.).
Authorship
Contribution: Q.S., C.V.C., and G.E.G. designed and conducted the research, analyzed data, and wrote the manuscript; and Y.C., P.T.S., F.X., and X.M.L. performed research and analyzed data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Qizhen Shi, Department of Pediatrics, Medical College of Wisconsin, 8701 Watertown Plank Rd, Milwaukee, WI 53226; e-mail: qshi@versiti.org; Christopher V. Carman, Harvard School of Public Health, 677 Huntington Ave, Boston, MA 02115; e-mail: ccarman@hsph.harvard.edu; or Gary E. Gilbert, VA Boston Healthcare System and Harvard Medical School, VA Medical Center–Boston, 9th Floor, Room 9D-136, 150 S Huntington Ave, Jamaica Plain, MA 02130; e-mail: gary_gilbert@hms.harvard.edu.

